4 Physical and Chemical Properties
This section provides information about available physical and chemical properties of PFAS. Understanding of the physical and chemical properties of PFAS is important for the prediction of their fate and transport in the environment. The available information about physical and chemical properties varies between the different PFAS. Tabulated values are included in the Physical and Chemical Properties Table (Table 4-1) that is provided as a separate Excel file. The Priority Topics for Fate and Transport, Surface Water, Source ID, and Site Characterization include information about perfluoroalkyl acid (PFAA) precursor biotransformation (see Section 1.3.3). In addition, a new tab named Biotransformation was added to the Physical and Chemical Properties Table (Table 4-1). The biotransformation table includes a compilation of available precursor kinetics data (see Section 1.3.3.2).
4.1 Challenges and Limitations Related to PFAS Physical and Chemical Properties
Understanding the physical and chemical properties of PFAS is important for the prediction of their fate and transport in the environment. More specifically, reliable values are very important when attempting to explain the environmental behavior of PFAS through mathematical fate and transport modeling, where small variations in values can have large implications on predictions (see Section 10.4 on data analysis and interpretation, which includes a discussion of fate and transport modeling). There is large variation in both the quantity and robustness of published data (a lot is known about some PFAS, but almost nothing about others) on chemical and physical properties of PFAS. Reliable physical and chemical properties of PFAS are scarce (for example, vapor pressure and Henry’s Law constants), and some of the available values are estimated using predictive mathematical techniques, which predict properties of compounds from knowledge of their chemical structure. These are collectively referred to as quantitative structure-activity relationship (QSAR) models. Predicted, as opposed to directly measured, values are accompanied by additional uncertainty that may be significant in certain fate and transport modeling scenarios. In addition, many of the available properties are based on the acid form of the PFAAs, which is not present in the environment except at low pH. These uncertainties can result in wide ranges of reported values and thus limit confidence in the precision of current fate and transport models. The Physical and Chemical Properties Table (Table 4-1 provided as a separate Excel file) summarizes some of the physical and chemical properties that are available for PFAS. The object of this table is to serve as a starting point for the research and selection of relevant properties by the practitioner. Selection of appropriate physical and chemical parameters for a specific use is beyond the scope of this document.
4.2 Physical Properties
This section briefly describes some standard physical properties of PFAS. Additional references for more information are provided. In addition, the Physical and Chemical Properties Table (Table 4-1 provided as a separate Excel file) summarizes some of the physical and chemical properties that have been published for PFAS and are discussed in this section.
4.2.1 Physical State/Appearance
Most PFAS are solids, often crystalline or powdery in form, at room temperature; however, shorter chained compounds (the acid forms of PFCA and PFSA, FTS and FTOH with a 4- to 6-carbon tail) tend to take liquid form at room temperature (melting point is addressed in Section 4.2.3).
4.2.2 Density
Density (ρ) is the mass per unit volume of a substance. For an individual PFAS compound (or mixture of PFAS) that exists as a liquid at ambient temperatures, density can influence its behavior in the environment.
If the density of the liquid PFAS is greater than that of water, the liquid PFAS has the potential to migrate downward through the water column in groundwater or surface water as a dense nonaqueous phase liquid (DNAPL); see ITRC (2003) for discussion on density and solubility impacts on DNAPL behavior. For example, 4:2 FTOH, which is a liquid at 20oC and has a density and aqueous solubility of 1.59 g/cm3 and 974 mg/L (Table 4-1), respectively, would be predicted to behave similarly to carbon tetrachloride (with a density of 1.59 g/cm3 and a solubility of 800 mg/L) if released into the environment as a pure product (also referred to as a neat liquid). However, if 4:2 FTOH dissolved in water, the change in density of the solution relative to water is unlikely to result in a separate layer. Floating separate-phase liquid layers have been observed on the surface of aqueous PFOA and PFOS solutions at high concentrations (Costanza et al. 2019). However, the formation of these layers appears to be driven by the tendency of PFAAs to accumulate and aggregate at air-water interfaces, and not by density. See Sections 4.2.8, 5.2.2.2, and 5.2.4.1 for additional information on the behavior of PFAS at air-water interfaces and in aqueous solutions.
4.2.3 Melting/Boiling Points
Melting and boiling point information refers to the temperature of phase transitions of pure compounds. These properties determine whether a specific pure PFAS compound will exist as a liquid, solid, or gas under typical environmental temperatures. These data can vary among references. Predicted melting and boiling point values are available for most PFAS, but empirically derived values are not available for many compounds. Predicted values are generally useful in understanding the various physical states of PFAS; however, the accuracy of these results is still unknown and warrants further investigation. Available data indicate that melting and boiling points of PFAS will tend to increase as the fluorinated chain increases in length. For example, the melting point of PFBA is -17.5°C while the melting point of perfluorotetradecanoic acid (PFTeDA) is 130–135°C. See Table 4-1 for a list of melting and boiling points.
4.2.4 Solubility
Solubility (S) refers to the ability of a given substance, the solute, to dissolve in a solvent. It is measured in terms of the maximum amount of solute dissolved in a solvent at a specified temperature and pressure. Typical units are milligrams per liter (mg/L) or moles per liter (mol/L). Currently, experimentally measured data for the solubility of PFAS in water are available for many of the well-studied compounds, but values are still needed for less studied compounds. This limited availability of experimental data should be considered when relying on PFAS solubility data. Most cited values are based on predicted or modeled values and the input values to these calculations may themselves be estimates. Further, because some PFAS can form different types of supramolecular assemblies, as discussed in Section 4.2.7, the reported water solubilities may include microdispersions of micelles or hemi-micelles in addition to truly solubilized molecules.
Values of solubility for the acid forms of PFAAs and other PFAS are summarized in Table 4-1. Reported values for solubility of individual PFAS analytes may vary depending upon the method used to determine solubility, the form of the analyte (that is, acid or salt), pH, salinity, and whether the value is empirical or obtained through modeling. For example, laboratory studies of water solubilities for PFOS indicate that solubility decreases when the water salt content increases (3M Company 2000). Other factors may affect the reported value of solubility as well.
4.2.5 Vapor Pressures (Vp)
Vapor pressure is an indication of the tendency of a substance to partition into the gas phase. Vapor pressure is a measure of volatility in that the higher the vapor pressure of a compound, the more volatile it is. Compounds with higher vapor pressures, because they are in the gaseous phase or sorbed to aerosols in the atmosphere, have a higher potential for long-range transport. Compounds with lower vapor pressures, which are more likely to remain in solid or liquid form, are transported only via soil or surface/groundwater (Barton, Botelho, and Kaiser 2008), unless they are dissolved into airborne water droplets or sorbed on airborne particulates, as discussed in Sections 5.3.2 and 6.1. Section 1.1.3.3 includes information about volatile PFAS. See also the PFAS and Vapor Intrusion Fact Sheet developed in 2025.
Values for vapor pressure of PFAS are summarized in Table 4-1. Although there are multiple values for PFCAs, FASEs, and FTOHs, very little data exist for measured vapor pressure values for PFSAs, fluorotelomer acids, and perfluoroalkyl ether compounds, and much of the data are extrapolated or modeled. Caution must be taken when using the vapor pressures for PFAAs listed in Table 4-1 with respect to the acid or anion form of the compound, which may have very different vapor pressures. Efforts were made to report the values for the acidic form in Table 4-1, but references are not always clear. These values also should not be used for their corresponding salt form for the same reason (for example, the vapor pressure of ammonium perfluorooctanoate has been measured experimentally to be three orders of magnitude lower than the vapor pressure of perfluorooctanoic acid at 25oC (Barton, Botelho, and Kaiser 2009)).
4.2.6 Henry’s Law Constant (Kh)
The Henry’s law constant (Kh ), as well as the air-water partition coefficient (Kaw), indicate the relative concentrations of a compound between an aqueous solution and gas phase at equilibrium (air-water distribution ratio) and provide an indication of the propensity of a chemical to remain dissolved in water versus volatilizing into the gas phase. A chemical with lower solubility and higher volatility will have a higher Henry’s law constant than a chemical with higher solubility and lower volatility. Section 1.1.3.3 includes information about volatile PFAS. See also the PFAS and Vapor Intrusion Fact Sheet developed in 2025.
For most organic compounds of moderate to low solubility, Kh can be approximated by:
Kh = (Vp)(M)/S
where Kh is the Henry’s law constant, Vp is vapor pressure, M is molecular weight, and S is solubility. This constant can be expressed in a variety of units or as the inverse (water-air distribution ratio); thus, the units of expression should always be confirmed prior to use of this constant. Kh also displays nonlinear temperature-dependence and is typically reported at 25oC, which is higher than most ambient environmental conditions.
Experimental and modeled Henry’s law constants are available for many PFAS in several families, including FTOHs (for example, Wu and Chang (2011) and Xie et al. (2013)), PFSAs, PFCAs, FTCAs, FTSAs, FASEs, and FASAAs (for example, Kwan (2001) and Zhang et al. (2010)). For PFAS that can dissociate into anions or cations, as discussed in Section 4.3.2, the Henry’s law constant is pH-dependent and reported constants may not be applicable depending on the pH conditions within the solution (for example, Rayne and Forest (2009) and Johansson et al. (2017)). In a study of airborne PFOA release in industrial settings, monitoring above sumps found concentrations 40 times greater when the pH was 1.8 than at neutral pH, and PFOA release from aqueous solution was found to be several times greater at pH 4 than at pH 7 (Kaiser et al. 2010). Henry’s law constant values, converted between a variety of units, are presented in Table 4-1. Corresponding Kaw values and, where available, the temperature and pH at which the value is relevant, are also presented.
4.2.7 Critical Micelle Concentration
Given the difference in behavior between the “head” and “tail,” traditional surfactants (surfactants that contain a hydrocarbon chain), when in water, tend to aggregate into micelles (form a sphere with the hydrophobic portion of the molecules on the inside) when present above a certain concentration. Surfactants can also form other supramolecular assemblies, such as hemi-micelles or mixed micelles (micelles composed of a mixture of surfactant-type molecules), either independently in solution or at boundaries occurring between phases (Krafft and Riess 2015).
Early studies of PFAAs concluded that they behaved like traditional surfactants and aggregated in both micelles and mixed micelles (Pedone et al. 1997; Downer et al. 1999). The theoretical threshold concentrations for aggregation, generally referred to as critical micelle concentrations (CMCs), for PFAS are presented in Table 4-1. However, there remain some observed properties of PFAS that do not fit the traditional understanding of micelle formation. For example, it appears that some reported CMC concentrations are above the known corresponding solubilities of that compound. For example, López-Fontán, Sarmiento, and Schulz (2005) reported a PFOA CMC of more than 12,000 mg/L when the reported solubilities of PFOA are generally less than 10,000 mg/L (see Table 4-1). Several reported CMC concentrations for PFOS are also above the reported solubility values (Yu et al. 2009; Sørli et al. 2020; Bhhatarai and Gramatica 2011). In addition, Costanza et al. (2019) reported a separate-phase liquid at the surface of high-concentration solutions of PFOS and PFOA. These anomalous breaks with traditional micelle formation behavior have yet to be fully explained in the literature. Some researchers have also postulated that the nature of PFAA supramolecular aggregations is much more complicated in environmental settings than formation of micelles at concentrations exceeding the CMC value, which is derived from single compound systems. Some have hypothesized that PFAA supramolecular aggregations may occur at concentrations much lower than the CMC in groundwater, due to interactions with particles and/or co-contaminants, formation of hemi-micelles, or spatially variable concentrations within soil matrices (Johnson et al. 2007; Yu et al. 2009). Other researchers have been studying the variable nature of PFAS micelles in the presence of solvents (Dong et al. 2021). These hypothesized supramolecular aggregations, coupled with the interrelationship of adsorption to interfaces such as the air-water interface, present an extremely complex set of interactions that may be occurring in the environment. It is evident that there is much more research to be done on this topic, which can aid our understanding in what may be a key PFAA characteristic that influences fate and transport in the environment. This topic is further discussed in Section 5.2.
4.2.8 Partitioning to Fluid-Fluid Interfaces
The amphiphilic structure of PFAAs (hydrophobic tail with hydrophilic head) implies that they behave like traditional surfactants, accumulating to the fluid-fluid interfaces (for example, air-water or NAPL-water) by orienting themselves along the air-water boundary so that their hydrophobic tails are in the air while their hydrophilic head groups are in the water (Krafft and Riess 2015; Figure 4-1). Although there has been a good deal of research done that confirms surface aggregation, the actual structure of the molecular assemblies has not yet been elucidated. Work done by Hasegawa et al. (2017), however, has provided some theories that the aggregational shape of PFAS may be more complex than that of traditional surfactants and depend on the length of the fluorinated tail. See Section 1.3.1.1 for information about air-water partitioning.
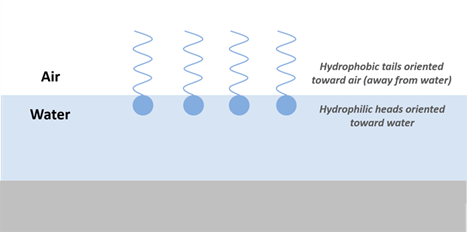
Figure 4-1. Example of expected orientation and accumulation of PFAS at air-water interface.
Source: D. Adamson, GSI. Used with permission.
Because of the low polarizability of the C-F bond relative to the C-H bond, PFAAs have a stronger affinity for interfaces than that of traditional hydrocarbon surfactants (Brusseau 2018). These strong surface-active properties and propensity toward self-assembly into films are what makes PFAAs extremely effective and widely used in a variety of applications such as water/grease repellent packaging and AFFF.
Studies have shown that PFAAs are enriched at the air-water interface up to eight times that of the bulk water concentration, with enrichment factors increasing with alkyl chain length (Psillakis et al. 2009; Costanza et al. 2019; Silva et al. 2019; Schaefer et al. 2019). A similar process is thought to occur for PFAAs at NAPL-water interfaces (Brusseau 2018; Silva et al. 2019; Brusseau 2020), however, the magnitude of accumulation at the NAPL-water interface appears to be far less than at the air-water interface at environmentally relevant concentrations. Recently, Costanza, Abriola, and Pennell (2020) reported that concentrations of PFOA, PFOS, and FOSA at the air-water interface were from 2 to 16 times greater than at the NAPL-water interface when the PFAS water concentrations were below 100 mg/L. This study also estimated that, in an unsaturated soil system with a dissolved water concentration below 1 mg/L, up to 87% of the PFOS mass accumulates at the air-water interface, while less than 4% of the mass occurs at the dodecane-water interface.
The degree of affinity that PFAAs exhibit for interfaces is related to the change in surface tension that occurs between the fluids when the compound is introduced to the system. Generally, the surface tension and affinity for the interface both go down as the amount of PFAA increases in the system, although the relationship is not linear. This means that the relationship is influenced by other factors that also effect the surface tension at interfaces (for example, the ionic strength and pH of the solution). For example, Constanza et al. (2019) found that the reduction in surface tension was more pronounced as the total dissolved solids increased (for TDS = 40, 400, and 1700 mg/L), which corresponded to greater accumulation of PFOA and PFOS at the air-water interface.
The adsorption coefficient for the fluid-fluid interfacial layer, termed the Ki (also called the Kai or Kia for the air-water interface or the Kni for the air-NAPL interface) is one metric for the affinity of a compound for fluid-fluid interfaces. However, calculation of the Ki is highly dependent on which assumed sorption model (Freundlich vs. Langmuir) is used, and values can vary by orders of magnitude at low concentrations (Schaefer et al. 2019). Because of this, it can be more useful to measure the affinity using the metric of surface excess (Γ, units of mg/m2 or mol/m2), which is the area-related concentration at the interface as compared to the bulk phase (Costanza, Abriola, and Pennell 2020). Because of the complicated nature of interfacial adsorption, adsorption coefficient values are not reported in Table 4-1.
4.2.9 Octanol/Water Partition Coefficient (Kow)
The Kow is defined as “the ratio of a chemical’s concentration in the octanol phase to its concentration in the aqueous phase of a two-phase system” (USEPA 2015). The Kow is a useful descriptor of the tendency of a compound to associate with hydrophobic or hydrophilic substances. Direct measurement of the Kow of PFAAs has proven difficult because they tend to aggregate at the interface between octanol and water (Kim et al. 2015), although some researchers have employed nontraditional methods with some success (Jing, Rodgers, and Amemiya 2009). Alternatively, Kow can be estimated using quantitative tools that predict physical and chemical properties. The Kow values that are typically tabulated for the PFCAs and PFSAs are for the acid form and are therefore not directly relevant to the anionic forms of PFCAs and PFSAs that exist within the typical range of environmental pH, although the Kow for the anionic form can potentially be estimated from the Kow for the neutral species (Escher et al. 2020).
Kow values are often used as surrogates for other properties, such as estimating Koc, the soil organic carbon/water partition coefficient; bioaccumulation; uptake in biological systems; and ecotoxicity. However, in the case of PFAS, the use of their Kow values as surrogates is generally not appropriate because PFAS do not behave like other well-researched nonionic polar chemicals. For example, the use of Kow as a surrogate for bioconcentration assumes partitioning to lipids in fatty tissues, but ionic PFAS such as PFOA and PFOS generally bind to proteins. See Section 5.5 for further discussion about PFAS uptake into aquatic organisms. For these reasons, Kow values are not presented in Table 4-1.
4.2.10 Organic Carbon/Water Partition Coefficient (Koc)
Koc is a metric commonly used to quantify the potential of a given dissolved compound to associate with, or sorb to, organic matter occurring in soil. Although short- and long-chain PFAAs both exhibit high potential mobility in water, long-chain PFAS are more likely to partition to sediment than short-chain PFAAs (Dalahmeh et al. 2018). Relative to commonly researched PFAS like PFOA and PFOS, studies have suggested Koc can be appropriately defined as a distribution coefficient (Kd) normalized to organic carbon content, thus implying Koc specifically represents the singular process of hydrophobic interaction (Milinovic et al. 2015). Broader reviews highlight the complexity and variability of processes that may contribute to the sorption of PFAS and significant differences between laboratory- and field-scale results (Li, Oliver, and Kookana 2018). These reviews, which cite the influence of pH, ionic composition, and other soil properties on sorption, suggest that simple hydrophobic interaction-based relationships may be unreliable for predicting partitioning for PFAS (see Section 5.2 for more discussion on partitioning). As such, the current state-of-science supports Koc being reported in relatively broad ranges on a compound-specific basis. The Koc values included in Table 4-1, while not an exhaustive list, are an indicator of the number of values currently available for PFAS. Discussion of the use of Koc, including site-specific Koc, in the prediction of PFAS transport in remedial scenarios is included in Section 10.4.2.
4.2.11 Partitioning to Biota
The extent of accumulation of PFAAs in organisms is controlled by several mechanisms, including uptake from food and from water, depuration, growth dilution, and biotransformation. These processes are described in Section 5. Within organisms, PFAA accumulation differs among tissues, depending on transport factors (for example, the ability to cross the blood-brain barrier, as discussed in Dassuncao et al. (2019) or active cellular transport (as discussed in Section 17.2.3) as well as partitioning. Partitioning is the subject of this section.
Similar to solids partitioning in the environment, the surfactant structure of PFAAs determines the types of interactions that occur within biota. Nonionic hydrophobic chemicals are known to partition into lipids within body compartments. PFAAs are amphiphilic, however, and thus are likely to interact with other organic molecules with both polar and nonpolar regions within living organisms. Many studies have found significantly different PFAA concentrations between body compartments, with the most accumulation occurring in the blood plasma, liver, and kidneys of many organisms (Labadie and Chevreuil 2011; Goeritz et al. 2013; Murakami et al. 2011). This marked difference in PFAA concentrations between bodily compartments has prompted some researchers to deem PFAAs “proteinophilic” to contrast with the description of other well-researched neutral hydrophobic environmental pollutants as “lipophilic” (Kelly et al. 2009; Labadie and Chevreuil 2011). Indeed, these differences in accumulation are likely, at least in part, due to interaction with/partitioning to proteins, which have binding pockets with varying charges that can interact with amphiphilic molecules like PFAAs. However, because the size, shape, and charge of binding pockets vary widely between proteins, there is an evident difference in PFAA binding between types of protein. Multiple studies have found strong interaction of PFAAs to serum albumin, fatty acid–binding proteins, and organic anion transporters (Chen and Guo 2009; Zhang, Ren, and Guo 2013). The magnitude of protein binding affinity differs with the type of PFAAs; Zhang, Ren, and Guo (2013) found that PFAA chain length and functional group determined the amount of binding affinity, but the relationship between binding and chain length was not linear. They also found that FTOHs (neutral charge molecules) did not bind to fatty acid–binding proteins, indicating that the charge of the functional group plays a large part in the protein interaction.
In addition to proteins, PFAAs likely interact with phospholipids (the basic building block of cell membranes). Dassuncao et al. (2019) found a positive correlation between PFAA and phospholipid content in several body compartments of pilot whales. It has been suggested that the interactions occur because of the amphiphilic nature of both phospholipids and PFAAs, which have the potential for binding at both their polar and nonpolar regions (Armitage, Arnot, and Wania, 2012). However, researchers have not found evidence that these interactions are the main determinant of tissue binding. Dassuncao et al. (2019) noted variability in the correlations between phospholipid content and tissue concentrations and concluded that the variability was likely due to the combined effect of protein and phospholipid partitioning.
For nonionic hydrophobic compounds, differences in lipid content can explain some of the variation in chemical concentration among organisms; under some conditions, individuals with higher lipid content have higher body burdens. In general, protein content differs to a much smaller extent among organisms than does lipid content, suggesting that differences in protein partitioning among individuals do not contribute strongly to differences in body burden among individuals. However, the sources of interindividual variation in PFAA body burdens is not well-understood and there is no current framework to incorporate our understanding of PFAS partitioning (for proteins and phospholipids, as well as lipids, which may still have a role) into reliable models for the prediction of bioaccumulation, biotransfer, or biomagnification.
4.3 Chemical Properties
This section briefly describes some standard chemical properties of PFAS. Additional references are provided for more information. In addition, the Physical and Chemical Properties Table (Table 4-1 provided as a separate Excel file) summarizes some of the physical and chemical properties that are available for PFAS.
4.3.1 Carbon-Fluorine (C-F) Bond Properties
The properties of PFAS are principally due to the unique properties of the carbon-fluorine bond. Some key chemical properties of this bond and the characteristics they impart to PFAS are provided in Table 4-2.
Table 4-2. Fluorine characteristics
| Fluorine Characteristic | Description | Effect | Resulting Property of PFAS |
|---|---|---|---|
| High electronegativity | Tendency to attract shared electrons in a bond | Strong C-F bond | Thermal stability |
| Chemical stability (low reactivity) | |||
| Polar bond with partial negative charge toward F | Strong acidity (low pKa)1 | ||
| Low polarizability | Electron cloud density not easily impacted by the electric fields of other molecules | Weak intermolecular interactions | Hydrophobic and lipophobic surfactant properties2 |
| Low surface energy | |||
| Small size3 | Atomic radius of covalently bonded fluorine is 0.72 Å | Shields carbon | Chemical stability (low reactivity) |
| 1When paired with an acid functional group such as a carboxylic or sulfonic acid 2When paired with a functional group that is hydrophilic (for example, a carboxylate) 3Smallest of the halogen atoms Å = angstrom |
|||
Properties such as the high electronegativity and small size of fluorine lead to a strong C-F bond, the strongest covalent bond in organic chemistry (Kissa 2001). The low polarizability of fluorine further leads to weak intermolecular interactions, such as Van der Waals interactions and hydrogen bonding (Kissa 2001). These unique properties of fluorine give many PFAS their mutually hydro- and lipophobic (stain-resistant) and surfactant properties and make them thermally and chemically stable. But not all of these characteristics (for example, surface activity) are universal to all PFAS.
4.3.2 Functional Group Properties
PFAS functional groups include carboxylates, sulfonates, sulfates, phosphates, amines, and others, as introduced in Section 2.2.2. These functional groups, including dissociated and undissociated forms, govern many fate and transport properties of PFAS. The ionic state of a compound determines its electrical charge and its physical and chemical properties, which in turn control its fate and transport in the environment. For example, the state (anionic or undissociated acid) of a given PFAS may alter aspects such as volatility and bioaccumulative potential. As further described below, due to their low acid dissociation constants (Ka), PFAAs are found in the environment in the anionic (negatively charged) state, except in very rare situations (for example, pH <3).
Functional groups of some PFAS (ionic PFAS) can dissociate into anions or cations in aqueous solution under appropriate pH conditions. For example, as discussed in Section 2.2.2, PFOA dissociates into the perfluorooctanoate anion and the hydrogen ion when dissolved in water over a wide range of pH conditions. The ion associated with the fluoroalkyl portion of ionic PFAS can be a negatively charged anion, a positively charged cation, or a zwitterion. Therefore, PFAS can be divided into four classifications based on functional groups (examples of the structures of many ionic PFAS can be found in Barzen-Hanson et al. (2017):
- anionic–contains one or more acidic functional groups such as carboxylic acids, sulfonic acids, sulfates, and phosphates, and can release a hydrogen ion, thereby forming an anion (see Figure 2-8 for PFBA dissociation)
- cationic–contains one or more basic functional groups such as amines, which can gain a hydrogen ion and form a cation, or have a permanent charge as in the case of a quaternary ammonium group
- zwitterionic–contains two or more functional groups, at least one of which can form an anion and one of which can form a cation
- nonionic–does not dissociate into ions; for example, alcohols.
Based on the behavior of other cationic and anionic surfactants, cationic PFAS are expected to have different environmental transport characteristics than anionic PFAS (Place and Field 2012). For example, sorption of organic anions such as PFAA anions is typically suppressed at higher pH due to electrostatic repulsion with the increasingly negative charge from deprotonated oxides and other functional groups present on the soil surface (Lee and Mabury 2017). Cations can be expected to sorb strongly to soils, which often possess a net negative charge over a range of environmentally relevant pHs. For example, cationic fluorotelomer-based PFAS in an AFFF product have been found to sorb strongly to soils and sediments (Barzen-Hanson et al. 2017). Zwitterionic PFAS can be expected to sorb to soils and sediment more strongly than anionic PFAS, but less strongly than cationic PFAS, owing to the mixed charges on the functional groups. The transport characteristics of specific PFAS are also highly dependent on matrix interactions, and detailed site-specific information is necessary to accurately predict PFAS transport (Guelfo and Higgins 2013). See Section 5.2.3 for further discussion of partitioning onto solid matrices.
For acids such as PFAAs, the acid dissociation constant Ka is the equilibrium constant for the dissociation of the acid in aqueous solution into the anion and hydrogen ion, and at dilute to moderate concentrations, is defined by the equation:
Ka = [anion–]*[H+]/[acid]
where [acid] is the concentration of the undissociated acid form, [anion–] is the concentration of the anion, and [H+] is the concentration of the hydrogen ion at equilibrium.
The dissociation constant is also commonly expressed as its negative logarithm, pKa, where:
pKa = – log10(Ka)
Higher pKa values indicate that an acid will dissociate less in water at a given pH than will an acid with a lower pKa. When the pH of a solution equals the pKa for a particular constituent, then one half of the constituent molecules will exist as the undissociated acid and one half will exist as the dissociated anion. PFAS with pKa values of 4 or less will exist in aqueous solutions at neutral pH (7) almost entirely as the dissociated acid (see Figure 4-2 for a representation of reported pKa values for PFOA in relation to environmental pHs). Because the undissociated acid and anionic forms of PFAAs may have very different physical and chemical properties, it is essential to distinguish between the undissociated acid form and the anionic form to select the appropriate physical and chemical parameters for fate and transport evaluations.
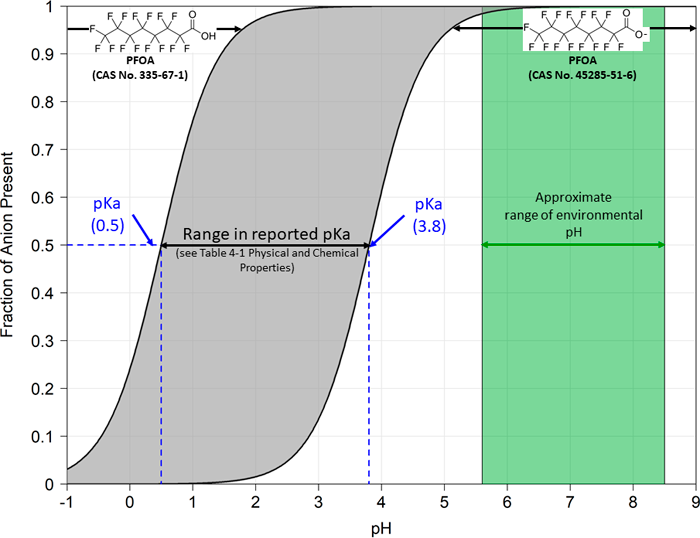
Figure 4-2. Titration curve for PFOA—relation of pKa to environmental pH.
Source: E. DiFilippo, S.S. Papadopulos & Associates, Inc. Used with permission.
Table 4-1 presents pKa values for PFAS. Limited model-predicted and experimental values are available for most PFAAs.
4.3.3 Thermal Stability
Thermal stability, the degree to which a chemical remains intact under thermal stress, is an important property to predict how long a chemical will persist in the environment. PFAAs, such as PFOA and PFOS, are extremely stable, thermally and chemically, and resist degradation and oxidation. Thermal stability of PFAAs is primarily attributable to the strength of the C-F bond in the fluoroalkyl tail (Kissa 2001), but the relative stability is also determined by the specific functional group that is attached to the fluoroalkyl tail. PFCAs and PFSAs are the most thermally stable fluorinated surfactants. The temperatures at which PFAAs decompose and complete mineralization occurs is an area of active research (see Section 12.4). Reports on temperature needed to destroy PFAS vary, but it seems that to destroy PFAS in soil, temperatures upwards of 1,000oC may be required (Colgan et al. 2018; Winchell et al. 2021). It was previously reported that limited PFOS, PFOA, and PFHxA mineralization (less than or equal to 72%) may occur at temperatures of 700oC (Watanabe et al. 2018). In this same study, mineralization reportedly increased to 90% in the presence of granular activated carbon and sodium hydroxide. The thermal stability is lower for the salts of PFAA compounds and depends on which cation is the counterion. For example, the 20% decomposition temperature of sodium perfluorooctanoate is 298°C, but it is 341°C for lithium perfluorooctanoate (Kissa 2001). Additionally, salts of PFSAs are more thermally stable than the corresponding salts of PFCAs (Kissa 2001).
4.3.4 Chemical Stability
Like thermal stability, knowledge of the chemical stability of a molecule helps predict its persistence in the environment. PFCAs and PFSAs have been shown to be persistent in the environment. PFCAs are resistant to oxidation under environmental conditions; however, transformation has been demonstrated in the presence of oxidants under extreme pressure. In contrast, transformation of precursors can be associated with substantial changes in the physicochemical properties of those compounds (CONCAWE 2016). See Section 1.3.3 for information about PFAA precursor biotransformation.
In the perfluorinated tail of the alkyl acids, the strength of the C-F bond, shielding of carbon by fluorine, and inductive effects (caused by fluorine electronegativity) also lead to chemical stability. For example, electron-rich chemical species called nucleophiles normally would be attracted to the partial positive charge of carbon. If these nucleophiles could get close enough to the carbon to bond, the subsequent reaction could replace a fluorine with the nucleophile and potentially make the molecule vulnerable to degradation. But the relatively large size of the fluorine atoms surrounding the carbon (when compared to hydrogen) prevents this from happening (Schwarzenbach, Gschwend, and Imboden 2003). This is why processes such as hydrolysis, which involve eliminating one or more fluorines, are ineffective at degrading the perfluorinated tails of PFAAs. Similarly, many PFAAs are resistant to degradation by oxidative processes that rely on a loss of electrons (Kissa 2001). PFAAs are also resistant to reductive processes, which involve gaining electrons. Despite having a high affinity for electrons, fluorine does not have vacant orbitals favorable for accepting additional electrons (Park et al. 2009). In contrast to the stability of perfluorinated tails, polar regions of PFAS (the functional groups), as well as polyfluorinated groups, can be vulnerable to a range of chemical transformations. See Section 5.4 for further discussion of abiotic and biotic transformations.
–
Updated January 2026.