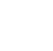11 Sampling and Analysis
Due to the extensive use of a wide array of PFAS resulting in trace levels of PFAS in most environmental media across the globe and the low parts–per–trillion screening and action levels, all aspects of a sampling and analysis protocol require a heightened level of rigor to avoid cross-contamination and achieve the level of accuracy and precision required to support defensible project decisions. This section focuses on providing the user with the appropriate tools and information to develop a site-specific sampling and analysis program to satisfy the project data quality objectives (DQOs). Accurate and representative data support the development of a defensible conceptual site model (CSM), and ultimately the final remedy.
| Section Number | Topic |
|---|---|
| 1.5 | Priority Topics for Sampling and Analysis (2025) |
| 11.1 | Sampling |
| 11.2 | Analytical Methods and Techniques |
| 11.3 | Data Evaluation |
| 11.4 | Source Identification |
Information on sample collection for PFAS is sparse, with only a handful of guidance documents available for a practitioner to reference. However, there are two peer-reviewed studies (Denly et al. 2019; Rodowa et al. 2020) on the potential for cross-contamination from commonly used sampling materials. These studies indicate that the potential for cross-contamination from the equipment used or possession of items that could potentially contain PFAS (for example, bug spray, food wrappers, and sunscreen) is extremely low and difficult to document. As of the date of this publication, most guidance documents default to a conservative approach due to a lack of information at the time of publication. Although the actual methods of sample collection are similar to those used for other chemicals, there are several considerations for the practitioner when establishing a sampling program for PFAS. These include selection of proper personal protective equipment (PPE), documentation of protocols for sample handling and decontamination procedures, use of nonbiasing material (for example, tubing, sample bottles, pumps) that will or could reasonably come into contact with the sample media, and implementation of quality control (QC) protocols to meet project DQOs, among other considerations. This section will give practitioners the tools needed to prepare a sampling program that adequately addresses project-specific DQOs and limits, to the extent practicable, potential cross-contamination and sources of potential bias.
Analytical methods are still evolving for PFAS analysis, with several in development (USEPA 2024). Although some methods have been published (PFAS Analytical Methods provided as an Excel spreadsheet), not all are discussed in this document because details included in these methods are subject to change prior to the methods being finalized.
The USEPA Office of Water has validated and published sample preparation procedures and analysis procedures applicable to groundwater, surface water, wastewater, landfill leachate, soil, sediment, biosolids, and tissue in EPA Method 1633A (USEPA 2024).
The USEPA Office of Land and Emergency Management has validated and published sample preparation procedure and analysis procedures applicable to groundwater, surface water, and wastewater. USEPA SW-846 Method 3512 is a sample preparation method, and USEPA SW-846 Method 8327 is the associated analytical method.
Three USEPA methods are validated and published for the analysis of PFAS in drinking water: USEPA Method 537 (USEPA 2009), USEPA Method 537.1, Version 2.0 (USEPA 2020), and USEPA Method 533 (USEPA 2019). For simplicity in the text, subsequent references to USEPA Method 537.1 assume the most current version. These methods are required for PFAS analyses of drinking water and include performance data for drinking water from surface water and groundwater sources. These methods are prescriptive in that changes to preservation (including sample collection, in the case of USEPA Method 537.1), sample extraction steps, and quality control requirements are prohibited by the methods.
The DOD has also validated and published a method (DOD AFFF01) for the determination of PFOA and PFOS in AFFF concentrates for demonstration of compliance to MIL-PRF-24385 (Willey 2021).
This section will be updated as new information on sampling considerations and analytical methods/procedures becomes available.
11.1 Sampling
11.1.1 General
Sampling conducted to determine PFAS concentrations in drinking water, other aqueous media, soil, sediment, air, biota, and other media is similar to that for other chemical compounds, but with several additional specific considerations and protocols. Typical guidance and procedures, such as ASTM International D 4823-95 and D 4448-01, USEPA compendium EPA 540/P-87/001a, OSWER 9355.0-14, USEPA SESDPROC-513-R2, and USEPA SESDPROC-305-R3, remain the basis for a PFAS sampling protocol. Because of the need for very low laboratory quantitation limits and the widespread use of PFAS in common materials, field and equipment blanks are needed in greater amount and frequency than in other analyses. In addition, special consideration may be needed to address the potential for background sources of PFAS in the environment, sample cleanup criteria, and the potential need for modified decontamination measures.
Examples of USEPA region-specific or program-specific PFAS sampling protocols include:
- USEPA (2019) Region 4, Laboratory Services and Applied Science Division, Athens, GA, Field Equipment Cleaning and Decontamination at the FEC, ASBPROC-206-R4, 2019
- DOD AFFF01 Determination of Perfluorooctanoic acid and Perfluorooctanesulfonic acid in Aqueous Film Forming Foam (AFFF) for Demonstration of Compliance to MIL-PRF-24385 (Willey 2021)
- State guidance:
- MA DEP (2023) Massachusetts Department of Environmental Protection, Sampling and Analysis for PFAS at Disposal Sites Regulated under the Massachusetts Contingency Plan.
- Washington Department of Ecology (2017) Quality Assurance Project Plan; Statewide Survey of Per- and Poly-fluoroalkyl Substances in Washington State Rivers and Lakes.
- NH DES (2022) New Hampshire Department of Environmental Services, Laboratory Testing Guidelines for Per- and Polyfluoroalkyl Substances (PFAS) at Waste Sites,
- MPCA 2022 Minnesota Pollution Control Agency, Guidance for Per- and Polyfluoroalkyl Substances (PFAS): Sampling
- Michigan Department of Environment, Great Lakes, and Energy (EGLE) PFAS Sampling Guidance Documents including:
- MI EGLE (2024) PFAS Sampling Guidance
- MI EGLE (2019) Surface Water Foam PFAS Sampling Guidance
- MI DEQ (2018) Wastewater PFAS Sampling Guidance
- MI DEQ (2018) Surface Water PFAS Sampling Guidance
- MI DEQ (2019) Fish Tissue Sampling Guidance
The list provided above is not comprehensive; project teams should consider state-specific and USEPA guidance as available.
A work plan (that is, a quality assurance project plan (QAPP)) should be implemented to address PFAS-specific considerations. If a work plan is not created for a project, the sampling and quality control (QC) elements outlined in Section 11.1.6 should be implemented. If regulatory procedures, methods, or guidelines are inconsistent with the needs of a PFAS sampling program, such as requiring the use of fluoropolymer tubing, the governing agency should be contacted directly to determine if an exception can be made or an alternate approach is needed. A CSM (if available, see Section 10.2) should be evaluated to support development of the project-specific DQOs as part of the work plan. The review should include information on previous site uses, PFAS use/manufacturing/handling practices, other possible contaminants and their uses, and/or related remediation activities and historical PFAS data, to determine all possible source areas of PFAS. Previous or ongoing remediation of other contaminants of concern can add a layer of complexity to a site’s geochemistry and the fate and transport of PFAS.
Although some sampling elements (for example, sample bottle, preservation, and holding times) are defined by analytical methods, these methods do not provide all the information that is needed to conduct a sampling event for PFAS. However, DOD AFFF01 contains the information needed to conduct sampling of AFFF concentrates. Tables 11-2 and 11-5 are included in the PFAS Analytical Methods Excel File, and cover the sample container types, sample size, number of containers required, and holding time and preservation requirements for each of these published PFAS analytical methods, respectively.
Communication with the laboratory before, during, and after sampling is conducted is critical in ensuring that project needs are met. If a sample is from an area known or suspected to be highly contaminated with PFAS, it is important that this is communicated to the laboratory. The chain-of-custody form should indicate samples that potentially contain a high concentration of PFAS. As stated in USEPA Method 1633A (USEPA 2024), laboratories should prescreen all samples to select the necessary sample preparation procedures and to avoid contamination of their laboratory equipment and contamination of other field samples.
Any water used for field QC blanks (for example, field and decontamination blanks) should be supplied by the laboratory performing the analysis. The laboratory should provide documentation verifying that the supplied water is PFAS-free. “PFAS-free” is the project-defined concentration that associated blank concentrations must be below (examples: less than the detection limit; less than half the limit of quantitation (LOQ)) to ensure an unacceptable bias is not introduced into the sampling and analysis processes and project data quality objectives can be met. The work plan should clearly state the project’s definition of “PFAS-free.” Review of the laboratory’s standard definition of “PFAS-free” upfront is necessary to ensure that it meets project needs and is a critical step in laboratory selection for a project. Laboratories verify the PFAS content of each batch of supplied water through analysis. Documentation of this verification should be maintained for data validation purposes and should be reviewed by the project team to ensure that the project’s definition of “PFAS-free” is met. If the water was not supplied by the laboratory, a sample of the water used in the field should be sent to the laboratory for testing.
11.1.2 Equipment and Supplies
Many materials used in environmental sampling can potentially contain PFAS. There is published research or guidance on how certain materials used by field staff or in sampling equipment affect sample results (see Denly et al. 2019; Field et al. 2021, Assessing the Potential for Bias in PFAS Concentrations during Groundwater and Surface Water Sampling, SERDP Project ER19-1205; Rodowa et al. 2020). However, a conservative approach is recommended to exclude materials known to contain the PFAS that are the target of the analysis from a sampling regimen, and such an approach should be documented accordingly in the work plan. Obtain and review all Safety Data Sheets (SDSs) before considering materials for use during PFAS sampling, as product manufacturing formulations can change over time. If PFAS or the terms “fluoro” or “halo” are listed on the SDS, it is recommended that piece of equipment/supply not be used. Exclusion from the SDS does not necessarily mean the equipment/supply is not contaminated with PFAS. PFAS may not have been used as a component of the equipment/supply, but as a material used in the manufacturing process itself (for example, mist suppressant or mold coating), which can result in the manufactured equipment/supply containing PFAS. If necessary, materials in question can be sampled and analyzed for PFAS, or thorough decontamination and collection of equipment blanks can provide sufficient quality assurances. Ultimately, a sampling program should produce defensible data, and the best way to protect the integrity of samples is to ensure they are not compromised by contaminants originating from sampling equipment or otherwise.
Due to the extensive use of a wide array of PFAS, sampling crews should review all materials and sampling protocols to avoid contamination and possible sorption issues. Examples of materials that, if used and contacted, could potentially introduce bias to samples include, but are not limited to:
- polytetrafluoroethylene (PTFE)
- waterproof coatings containing PFAS
- fluorinated ethylene-propylene (FEP)
- ethylene tetrafluoroethylene (ETFE)
- low-density polyethylene (LDPE)
- polyvinylidene fluoride (PVDF)
- pipe thread compounds and tape.
A tiered approach should be implemented for materials restrictions, where the first tier would include restrictions on the sampling materials that will come in direct contact with the sample media, and the second tier would include restrictions on what materials are allowed on sampling personnel or within the staging area. The focus on restrictions within the second tier should consider how reasonable the potential for contact with the sample media is when good sampling practices are employed, the practicality of the restriction (for example, does it compromise employee safety or increase an exposure risk), and the documentability of the requirement (can the restriction or measure be properly documented). Program-specific sampling protocols such as those previously listed in this section often identify materials and equipment that can be used in PFAS-focused investigations, as well as materials that should be avoided because they are known or suspected to be potential sources of PFAS. However, as noted in peer-reviewed studies (Denly et al. 2019; Rodowa et al. 2020), the potential for cross-contamination from sampling materials that are not likely to contact the sample media or incidental contact (for example, rain splatter off a treated raincoat) is minimal, therefore, the focus should be on material in direct contact with the sample media and with a reasonable potential for contact. It is also recognized that a PFAS-free alternative may not be readily available or confirmable. In such instances a robust quality assurance (QA) program consisting of appropriate equipment blanks can be implemented to address this.
There may be instances when it is not possible to eliminate sampling materials that may affect sample results. For example, problematic materials may be needed at sites where co-contaminant or facility hazards warrant the use of PPE such as Tyvek suits or flame-retardant coveralls for worker safety. Additionally, this could be an issue in emergency response scenarios if a sampler needs to collect a sample before a proper program is established or before proper sampling materials can be obtained.
11.1.3 Bottle Selection
Sample container material recommendations are dependent on the analytical method. Containers should be supplied by the laboratory and laboratory-verified to be PFAS-free, as defined by the work plan. USEPA SW-846 Method 8327 was validated using polypropylene containers for groundwater, surface water, and wastewater sampling. However, USEPA SW-846 Method 8327 states that other types of containers such as high-density polyethylene (HDPE) may be used if the needs of the project can be met with their use. USEPA Method 533 and USEPA Method 537.1 require the use of polypropylene or other plastic containers, such as polyethylene, that meet the QC requirements (Section 11.1.7.1). USEPA Method 1633A requires the use of HDPE containers for wastewater, groundwater, surface water, landfill leachate, biosolids, soil, sediment, and tissue sampling.
The volume of aqueous sample that is required for analysis varies from method to method and the mass of solid material required for analysis is dependent on the matrix of the sample.
Non-potable water samples do not require a chemical preservative, unless otherwise required by the cited analytical method(s). Clean laboratory-provided HDPE or polypropylene bottles are recommended; typically, 125-mL to 1-L bottles may be used, but the sample volume may depend on the analytical method used.
In USEPA Method 1633A, samples are prescreened to determine if they contain high concentrations of PFAS analytes or other potential interferences (for example, landfill leachates) that would prohibit the whole aqueous sample from being prepared. For prescreening analysis, the method requires collection of an additional, smaller aliquot of wastewater, groundwater, or surface water. This second aliquot allows the laboratory to prescreen the sample without affecting the integrity of the sample collected for analysis. Coordinating with the laboratory is crucial to ensure an adequate number of each type of required sample container is provided for the field activity.
Best practices in sample preparation should be used when selecting the size, volume, and representativeness of samples. To minimize effects from analyte sorption on sample containers, USEPA Methods 537.1 and 533, and USEPA Method 1633A all require the laboratory to prepare the entire aqueous sample collected, including sample container rinsate(s). DOD AFFF01 requires the container holding the diluted AFFF concentrate be prepared in its entirety, including a rinse of the container.
11.1.4 Sample Preservation, Shipping, Storage, and Holding Times
Sample preservation, shipping, storage, and holding time requirements are dependent on the method used. Drinking water methods (USEPA Method 537.1 and 533) are the only USEPA methods requiring the addition of a chemical preservative at sample collection. USEPA Method 537.1 requires the addition of Trizma®, while USEPA Method 533 requires the addition of ammonium acetate, as detailed in section 8 of each of the methods. According to both methods, samples must be chilled during shipment and not exceed 10°C during the first 48 hours after collection. When they are received by the laboratory, samples must be at or below 10°C and stored in the laboratory at or below 6°C until extraction. These two methods differ in their required holding times, as USEPA Method 537.1 requires samples to be extracted within 14 days of collection, while USEPA Method 533 requires samples to be extracted within 28 days of collection.
The shipping, storage, and holding time requirements for wastewater, groundwater, and surface water samples stated in USEPA SW-846 Method 8327 differ than those provided by USEPA Method 1633A. USEPA SW-846 Method 8327 requires all samples and sample extracts to be chilled from the time of sample collection to analysis and not exceed 6°C. Since a holding time study was not performed in conjunction with the validation of USEPA SW-846 Method 8327, the method offers a holding time of 14 days from sample collection to sample extraction and a 30-day holding time from sample extraction to sample analysis as a guideline.
The shipping, storage, and holding time requirements contained in USEPA Method 1633A are based on a published holding time study for PFAS by Woudneh et al.2019, as well as the holding time studies conducted in conjunction with the single- and multi-laboratory validation studies for USEPA Method 1633 (Willey et al 2021, 2023, 2023, 2023, 2023). While the statistically determined holding time requirements are similar for each matrix, they are not identical (see Table 11-5, included in the PFAS Analytical Methods Excel File). Observed losses and/or gains in the concentration of some PFAS, most likely caused by transformation of precursors, resulted in shorter holding times for samples and extracts. As stated in Method 1633A (USEPA 2024) for aqueous samples, “conversion of certain perfluorooctane sulfonamide ethanols and perfluorooctane sulfonamidoacetic acids have been observed after 7 days. Specifically, NMeFOSE and NEtFOSE may undergo transformation to NMeFOSAA and NEtFOSAA respectively when stored at 0 – 6 ºC, but not when stored at or below -20 ºC. Therefore, if NMeFOSE, NEtFOSE, NMeFOSAA and/or NEtFOSAA are analytes of concern for a given permit or project, either store the samples at or below -20 ºC, or extract the samples within 7 days of collection.” Also, due to the observed extreme loss of NFDHA, an additional caveat in the method recommends samples be extracted and analyzed for solids and tissues as soon as possible if NFDHA is an important analyte for the project (USEPA 2024). This issue was addressed by the DOD Environmental Data Quality Workgroup in a March 2025 memo when using 1633A for PFAS investigations (USDOD 2025).
11.1.5 Decontamination Procedures
Sampling equipment should be thoroughly decontaminated before mobilization to each investigation area and between sample locations at each investigation area or as required in the site-specific work plan. Field sampling equipment, including oil/water interface meters, water level indicators, nondisposable bailers, and other nondedicated equipment used at each sample location requires cleaning between uses. The SDSs of detergents or soaps used in decontamination procedures should be reviewed to ensure fluorosurfactants are not listed as ingredients. Use laboratory-verified PFAS-free water for the final rinse during decontamination of sampling equipment. Decontaminate larger equipment (for example, drill rigs and large downhole drilling and sampling equipment) with potable water using a high-pressure washer or steam. To the extent practical, rinse parts of equipment coming in direct contact with samples with PFAS-free water. Heavy equipment is best cleaned within a decontamination facility or other means of containment (for example, a bermed, lined pad and sump, or a portable, self-contained decontamination booth). Potable water sources should be sampled in advance and analyzed for PFAS, as well as during the sampling event to verify the water remained PFAS-free. If the potable water source does not meet the project-defined criteria for PFAS-free water, an alternate water supply should be considered. Wherever possible, rinse equipment with laboratory-verified PFAS-free water immediately before use.
An example of decontamination procedures as published in the Minnesota Pollution Control Agency “Guidance for Per- and Polyfluoroalkyl Substances (PFAS): Sampling” (MPCA 2022) is as follows:
- Equipment caked with drill cuttings, soil, or other material will initially be scraped or brushed. The scrapings will be collected, containerized, and disposed in accordance with government regulations.
- Equipment will then be sprayed with potable water using a high-pressure washer.
- Washed equipment will then be rinsed with laboratory-verified PFAS-free water.
- Decontaminated downhole equipment (for example, drill pipe, drive casing, bits, tools, bailers, etc.) will be placed on clean plastic sheeting (PFAS-free) to prevent contact with contaminated soil and allowed to air dry. If equipment is not used immediately, it will be covered or wrapped in plastic sheeting to minimize airborne contamination.
- Field sampling equipment and other downhole equipment used multiple times at each sample location will require cleaning between uses utilizing a four-stage decontamination process. The equipment will first be rinsed in a bucket containing a mixture of potable water and PFAS-free soap. The equipment will then be rinsed in each of two buckets of potable water. Water used for the final rinse during decontamination of sampling equipment will be laboratory-verified PFAS-free water.
Decontamination solutions should be replenished between sampling locations as needed. Spent decontamination fluids should be containerized, properly labeled, and appropriately disposed of as investigation-derived waste (IDW), based on plans included in the site-specific QAPP or work plan.
11.1.6 Field QC Samples
Field QC samples are a means of assessing quality beginning at the point of collection. Such field QC samples typically include field reagent blanks, source water blanks, equipment rinse blanks, and field duplicates. Collection and analysis of field QC samples are important for PFAS investigations because of very low detection limits and regulatory criteria (parts per trillion (ppt)), to ensure accuracy and representativeness of the results for the sampled media, and to assess potential cross-contamination due to the extensive use of PFAS. A sampling program should be designed to prevent cross-contamination and anthropogenic influence. However, the widespread commercial use (historical and current) of PFAS-containing products, and especially their prevalence in commonly used sampling materials and PPE, should inform the sampling program. PFAS sites may also have a wide range of concentrations with varying families of PFAS, as well as co-contaminants. Furthermore, PFAS sites have the potential to be high profile in nature. Therefore, a comprehensive site-specific QAPP or work plan addressing DQOs and field QC samples, including frequency, criteria, and procedures, is vital to a PFAS sampling program (see also Section 11.3, Data Evaluation).
When planning QC sample frequency, the risk of cross-contamination should be considered. Cross-contamination can occur from several sources, including field conditions, ineffective decontamination, incidental contact with PFAS-containing materials, and sampling equipment and materials that were manufactured alongside PFAS-containing equipment.
Of all the USEPA PFAS methods, only USEPA Methods 537, 537.1 and 533 contain specific requirements for the field QC samples that must be collected and accompany samples to be analyzed for PFAS. These include field duplicates, matrix spikes and duplicates, and a minimum of one field reagent blank for each set of samples per sampling site. USEPA Methods 537, 537.1 and 533 specify the frequency of the field duplicate or matrix spikes in terms of extraction batch (one per extraction batch, not to exceed 20 field samples), not collection frequency. Although the other USEPA PFAS methods do not contain any field QC sample requirements, Table 11-1 provides a list of field QC samples typical for the collection of these matrix types and their typical minimum frequency. Once field QC sample data are obtained, they should be evaluated against the field samples by a person knowledgeable on the DQOs set forth in the site-specific QAPP or work plan. For laboratory QC considerations, see Section 11.2, Analytical Methods/Techniques.
Table 11-1. Typical field QC samples
| QC Sample | Description | Minimum Suggested Frequency |
|---|---|---|
| Field reagent blank (FRB) | Laboratory-provided PFAS-free reagent water that, in the field, is poured into an empty sample bottle or a sample bottle containing only preservative (if required) | One per day per matrix per sample set |
| Source water blank | Water collected from potable water source that is used during the sampling processes (such as decontamination and drilling processes) | One per site, preferably prior to sampling event (if possible) and at least once during sampling event |
| Equipment rinse blank (ERB) | Final rinse sampling equipment with laboratory-verified PFAS-free water (decontamination blank); prior to the sampling event | One prior to the start of a sampling event per piece of sampling equipment following an initial decontamination
One per day per piece of sampling equipment used for each matrix sampled (during or at the end of the day) following decontamination |
| Field duplicate | Two samples collected at the same time and location under identical circumstances | One per day per matrix, or one per 20 samples per matrix, whichever is greater |
11.1.6.1 Field Reagent Blank
A field reagent blank (FRB), as described in USEPA Methods 537, 537.1 and 533 for collection of drinking water samples, consists of a sample bottle filled with PFAS-free reagent water prepared in the laboratory, sealed, and shipped to the sampling site along with the sample bottles. An empty sample bottle containing only preservatives (same as those used for the samples) is shipped along with each FRB into which the sampler pours the unpreserved PFAS-free reagent water contained in another bottle that was sent to the field and seals and labels the bottle for shipment along with the samples back to the laboratory for analysis. FRBs help to determine if PFAS were introduced into the samples during sample collection/handling and help to account for additional factors, such as introduction of contaminated airborne particles. A laboratory reagent blank is also analyzed in a laboratory setting to ensure that background contamination is not being introduced in the laboratory from reagents or water supplies.
FRBs may also be warranted during collection of sampling media other than finished drinking water. In lieu of using a prepared quantity of laboratory reagent water/preservative solution as for drinking water FRB, a field blank can be prepared in the field using laboratory-verified PFAS-free water (may be equivalent to the laboratory reagent water) and filling an empty sample container in the field, which is then sealed and labeled as a field blank. This sample will be analyzed in the same manner as the normal samples and can indicate whether or not PFAS were introduced during sample collection/handling, and help to account for additional factors, such as introduction of airborne particulates.
As discussed above, the frequency of FRB samples for finished drinking water sampling is one FRB for every sample set at each site. A sample set is described in USEPA Methods 537.1 and 533 as “samples collected from the same sample site and at the same time” (USEPA 2020; USEPA 2019).
11.1.6.2 Source Water Blank
Large quantities of water may be necessary to carry out a field sampling program for various reasons, including decontamination and certain drilling techniques (Section 11.1.5, Decontamination Procedures). Site groundwater or surface water should not be used as source water at sites that are under investigation for PFAS, as they have potential to contain PFAS. PFAS test results of publicly supplied water at a site should be evaluated when considering it for use as source water. Regardless, it is imperative that any water used as source water be sampled and analyzed. For equipment that may come into contact with samples of any media type, a multistep process is common to adequately prevent cross-contamination. Quantities of laboratory-verified PFAS-free water are generally limited and can be costly. Therefore, potable water sources are typically used in initial decontamination steps. It is imperative that these water sources be sampled and analyzed in the same manner as normal samples, prior to and even during a PFAS sampling program, to ensure that source water is not contributing to PFAS detections in normal samples. Further, many laboratories do not provide a verification of the laboratory-supplied DI water being PFAS-free unless specifically requested. Submission of a laboratory deionized water blank could be required based on DQOs of the specific project.
Collect a sample from the source the same way it is collected for use (for example, if the source water is collected through a hose, collect the source water blank from that same hose). If there are unnecessary fittings or hoses attached for collection of the source water, consider removing them for the duration of the sampling program to avoid contamination from PFAS that may be present in these materials.
Frequency of collection of such source water blanks is up to the professional judgment of the project manager, site owner, and other stakeholders and is detailed in the project-specific work plan. The source water should be sampled at least once prior to starting the field sampling program and once during the sampling event in case the analysis reveals that a different water source should be found. A more conservative sampling program may include provisions for additional periodic sampling in cases where the conditions of the source water may change.
11.1.6.3 Equipment Rinse Blank
Equipment Rinse Blanks (ERB) should be collected from nondedicated equipment/supplies at the start of the sampling event prior to sample collection to verify that nondedicated equipment/supplies are PFAS-free (Section 11.1.2). Thereafter, ERBs should be collected from nondedicated sampling equipment at a minimum rate of once per day (either during or at the end of each day) following decontamination and prior to its next use for the duration of the sampling event.
ERBs are those collected by rinsing a piece of field sampling equipment/supplies with laboratory-verified PFAS-free water and collecting the rinse water in a sample container for PFAS analysis. ERB collection is not required by USEPA Methods 537.1 or 533 because drinking water compliance samples are generally collected from the source without the use of other equipment. ERB collection for other programs is dependent on the sampling media and methods that are employed at a site. Generally, any equipment that is reused throughout the sampling program, or is nondedicated, and must be decontaminated, should have an ERB collected from it. That is, if a piece of equipment is decontaminated, an ERB should be collected from it after decontamination and prior to its next use. The frequency of collection of ERBs can be reduced by using all dedicated or disposable equipment where possible. However, many of these options are limited due to the extensive use of PFAS in many of these equipment materials. ERBs should also be considered for dedicated equipment prior to and during a sampling event if the PFAS contribution from equipment is unknown or suspected.
Field ERB collection frequency is largely up to the professional judgment of the project manager or other stakeholders and is dependent on the sampling media and methods and project-specific DQOs. For instance, collection of ERBs prior to initiating an event may be appropriate if rental equipment or subcontractor material that could have been used on another site is being used. The project team may also consider collecting an ERB from each piece of equipment used by the sampling team (ERB per sample setup) when multiple sampling setups are being implemented. Collection of an ERB from decontaminated soil sampling trowels may only warrant a frequency of once per day, whereas groundwater pumps may warrant an ERB prior to the pump being deployed down each well, due to their more rigorous decontamination procedure and higher contact time with the groundwater being sampled.
11.1.6.4 Field Duplicate
Field duplicate (FD) samples are two samples collected at the same time and location under identical circumstances and treated exactly the same throughout field and laboratory procedures. The analytical results from these identical samples are used to evaluate the precision of sample collection, preservation, storage, and laboratory methods.
USEPA Methods 537.1 and 533 do not specify the frequency of FD collection for finished drinking water samples; however, they do specify the frequency of preparation (once per extraction batch, not to exceed 20 field samples). A more conservative sampling program may indicate a frequency of one FD per 10 field samples per matrix. FD collection frequency should be discussed with stakeholders, as necessary, and be evaluated as part of the comprehensive site-specific QAPP or work plan.
11.1.6.5 Additional QC Samples
In addition to the field quality control samples described above, replicate volume or mass may be collected for split samples and/or matrix spikes to provide project managers additional quality assurance regarding identification of PFAS target analytes and precision and bias in measured sample concentrations. Split samples are defined here as co-located quality control samples, taken at the same time and each sent to a different laboratory. These types of samples do not apply to routine compliance monitoring situations and may not be required in all sampling events. Aqueous QC samples should not be split into two samples from the original container. Analysis of these QC samples provides a measure of interlaboratory variability.
A performance evaluation (PE) sample, which contains project analytes with known concentrations of PFAS, may be submitted to the laboratory as a blind sample. Analysis from this sample provides a positive control from a second source.
11.1.7 Sampling Procedures
The Priority Topics for Sampling and Analysis include information about leaching methods (see Section 1.5.1), fish sampling and laboratory homogenization (see Section 1.5.2), consumer product testing (see Section 1.5.4), ISM for PFAS sampling (see Section 1.5.5), and concrete and asphalt sampling and analysis (see Section 1.5.6).
Standard sampling procedures and practices can be used at most PFAS sites and only require extra care and attention paid to the types of materials used. However, there may be some exceptions and additional considerations related to PFAS behavior and issues associated with potential use of PFAS-containing or PFAS-adsorbing sampling equipment and supplies, as previously discussed. A site-specific QAPP or work plan must contain the standard operating procedures incorporating these considerations and regulatory agency or client requirements. Refer to Section 11.1.2 for materials to avoid during sampling and drilling. Consult the supplier to determine if PFAS-free options are available.
Pretesting any equipment or supplies to be used is ideal, although it is recognized that this is not typically feasible. As such, ERBs are recommended to ensure the decontamination of supplies (for example, bailers, pumps, beakers, and dippers) is effective.
In addition, the sampling team must document any observations during the sampling event that could be a source of bias (for example, the presence of PTFE tape on a faucet).
11.1.7.1 Drinking Water
Sampling a “potable water source,” as defined by the USEPA SDWA (Section 1401(4), August 1998), is conducted according to protocols established in USEPA Methods 537.1 and 533. These protocols define sample bottle preparation, sample collection, field reagent blanks, sample shipment and storage, and sample and extraction holding times. The drinking water source is further defined here as a public drinking water supply, as opposed to a private drinking water supply, as it applies to USEPA Methods 537.1 and 533. USEPA Methods 537.1 and 533 may also apply in instances when the water quality of the private drinking water supply source is similar to finished drinking water (for example, has low level of total organic content). The following summarizes the sampling considerations described in these protocols:
- For Methods 537 and 537.1: Sample bottle is a laboratory-provided 250-mL polypropylene bottle fitted with a polypropylene screw cap. For finished (treated) drinking water sampling only, a preservation agent is provided inside each bottle prior to sample collection. This agent acts as a buffer (Trizma®, pH 7, 5 g/L) and removes free chlorine from chlorine-treated drinking water supplies.
- For Method 533: Sample bottle is a laboratory-provided 100–250-mL polypropylene, or other plastic that meets method QC requirements, bottle fitted with a polypropylene screw cap. For finished (treated) drinking water sampling only, a preservation agent is provided inside each bottle prior to sample collection. This agent (ammonium acetate, 1 g/L) sequesters free chlorine to form chloramine in chlorine-treated drinking water samples. The sample handler must avoid PFAS contamination during sampling by thoroughly washing their hands and wearing nitrile gloves.
- Open the tap and flush the water (approximately 3–5 minutes) to obtain a “fresh” sample. Collect the sample while water is flowing, taking care not to flush out preservative. Samples do not need to be headspace-free. Cap the bottle and, if applicable, agitate to completely dissolve preservative.
- Keep sample sealed and place sample on ice for shipment.
- Samples must be chilled during shipment and must not exceed 10°C during shipment.
- Laboratory extraction of the sample must take place within 14 days of collection (USEPA Method 537.1) or within 28 days of collection (USEPA Method 533).
Based on a review of industry experience and guidance, additional considerations for collecting drinking water samples for PFAS analysis are as follows:
- The sample should ideally be cold water collected from a tap or spigot located at or near the wellhead or pump house and before the water supply is introduced into any storage tanks or treatment units. If the sample must be collected at a point in the water line beyond a tank, a sufficient volume of water should be purged to provide a complete exchange of fresh water into the tank at the tap or spigot. If the sample is collected from a tap or spigot located just before a storage tank, spigots located downstream of the tank should be turned on to prevent any backflow from the tank to the tap or spigot. Several spigots may be opened to provide for a rapid exchange of water. If collecting a sample to characterize human or other exposure, the sample should be collected from the tap or spigot at the point of use.
- When sampling from a drinking water well that is not in regular use, purge water until water quality parameters (that is, pH, specific conductance, dissolved oxygen, oxidation-reduction potential, turbidity, and temperature) have stabilized according to project-specific requirements, to ensure formation water (as opposed to stagnant well column water) will be sampled. An adequate purge is achieved when the pH and specific conductance of the potable water have stabilized (for example, within 10% across three consecutive measurements) and the turbidity has either stabilized or is below 10 nephelometric turbidity units. Note: According to USEPA (2013), pg. 21 “[a] well with an intermittently run pump should, in all respects, be treated like a well without a pump. In these cases, [water quality] parameters are measured and the well is sampled from the pump discharge after parameter conditions have been met. Generally, under these conditions, 15 to 30 minutes will be adequate.”
- When sampling from a tap, the tap must be protected from exterior contamination associated with being too close to a sink bottom or to the ground. Contaminated water or soil from the faucet exterior may enter the bottle during the collection procedure because it is difficult to place a bottle under a low tap without grazing the neck interior against the outside faucet surface. If the tap is obstructed in such a way that prevents direct collection into the appropriate sample container, it is acceptable to use a smaller container to transfer sample to a larger container. The smaller container should be made of HDPE or polypropylene and should be either new or decontaminated as specified in Section 11.1.5. Evaluation of the transfer container is recommended to ensure that it does not introduce a bias.
- When filling any sample container, care should be taken that splashing drops of water from the ground or sink do not touch either the interior of the bottle or cap.
- Leaking taps that allow water to discharge from around the valve stem handle and down the outside of the faucet, or taps in which water tends to run up on the outside of the lip, are to be avoided as sampling locations.
- Disconnect any hoses, filters, or aerators attached to the tap before sampling.
Taps where the water flow is not constant should be avoided because temporary fluctuation in line pressure may cause clumps of microbial growth that are lodged in a pipe section or faucet connection to break loose. A smooth flowing water stream at moderate pressure without splashing should be used. The sample should be collected without changing the water flow.
11.1.7.2 Groundwater
Groundwater is typically sampled from a well, and therefore specialized equipment is required. When constructing monitoring wells prior to groundwater sampling, care should be taken to ensure well-construction materials are free of PFAS. Radford et al. (2023) detected PFAS in coated and uncoated bentonite formulations with more individual PFAS present at higher concentrations in coated formulations. PFAS have also been added to various concrete formulations (Kissa, 1994). Drilling greases, grout, and all well construction materials should be confirmed to be PFAS-free prior to use. Additionally, water added to the formation for drilling purposes (mud mixtures, managing running sands, etc.) may need to be tested for PFAS prior to use and should be thoroughly developed out of the completed well prior to sampling. Some of the various types of purging and sampling equipment available for groundwater sampling are described in ASTM International Standard Guide for Sampling Ground-Water Monitoring Wells, D 4448-01 (ASTM 2007) or Compendium of Superfund Field Operations Methods (USEPA 1987).
Within the context of sample collection objectives outlined in a site-specific work plan, the sample depth selection should consider the potential stratification of PFAS within the aquifer, the potential for phase partitioning, and the tendency for PFAS to accumulate at the air/water interface. In addition to depth location within the water column, consideration should be given to the well construction, screened interval, and site geology to ensure that the well is representative of site conditions and all relevant chemicals of concern (COCs). For more information on phase partitioning, see Section 5.2.
The most inert material (for example, stainless steel, silicone, polypropylene, and HDPE), with respect to known or anticipated contaminants in the well(s), should be used whenever possible. Purging and sampling equipment could be manufactured from components that might include PFAS. For example, pump components, such as O-rings, gaskets, bladders, stopcocks, and wiring insulation, may contain PFAS. Contact between the groundwater, the sample, and PFAS-containing equipment provides opportunities for the sample to become contaminated and should be avoided when possible. In some cases, components that contain PFAS can be switched out for HDPE or polyethylene. Consult with the equipment vendor to determine if they have PFAS-free alternatives. In addition to equipment, ensure that field supplies that may contact the sample, such as tubing or bailer twine, are PFAS-free and will meet the DQOs. Dedicated sampling equipment installed in existing wells prior to PFAS investigations may contain components that are not PFAS-free, and the equipment’s chemical properties may be challenging to verify. It is best practice not to collect samples using dedicated equipment that may include PFAS-containing components. If samples are collected with dedicated equipment with some unknown components, PFAS detections should be evaluated in an uncertainty analysis.
Where sampling for co-contaminants requires use of PFAS-containing materials, sampling events should be separated to avoid contamination from these materials. The PFAS sampling event would be completed first, followed by the sampling event for the co-contaminants. In some cases, it may be acceptable to use the same equipment at a concurrent sampling event. For circumstances that warrant, such as very deep wells or sites with co-contaminants, samples may be collected in duplicate with and without existing dedicated equipment. If PFAS analyses show that the equipment does not impact results, the equipment may be kept and used long term. However, this determination is dependent upon project-specific requirements and should be allowed by a project manager only with full disclosure to all stakeholders. It may also be acceptable to simply collect an ERB after fully decontaminating equipment containing PFAS components to confirm it does not contribute to groundwater sample concentrations. A site-specific procedure should be outlined in the QAPP or work plan.
The practitioner should determine that the sampling method is compatible with the site DQOs, including whether the method:
- collects site-specific analytes of interest
- is effective at the expected concentrations
- provides sample volume that meets the lab method requirements
- minimizes turbidity
- minimizes purge water
- is likely to include equipment with PFAS-containing components that contact the sample
Low-flow or passive sampling techniques are preferred for collection of groundwater samples for PFAS to keep the turbidity of samples and purge-water volume to a minimum. PFAS are mobile in groundwater and have relevant transport pathways, including advection and diffusion, that are similar to those for other contaminants (Section 5.1.2) and conducive to passive sampling (Section 11.1.7.6) in groundwater. See Section 11.2.1.2 for issues associated with elevated levels of suspended solids in aqueous samples.
Bailers should be used with caution due to the potential for PFAS to accumulate at the air/water interface, because they may increase turbidity, and because of the potentially large volume of purge water produced. If bailers are used, it is important to make sure that at least three well volumes are purged to remove static surface conditions and potentially stagnant or aerated water. Volume-purge pump sampling for PFAS is discouraged because of the large amounts of purge water generated and the tendency to increase turbidity.
For background guidance on the use of passive sampling, consult the ITRC’s technical guidance document (ITRC 2024) which provides guidance on passive sampling technologies in aqueous environments (see also Section 11.1.7.6).
Samples should not be filtered, because filters may be either a source for contamination (Ahrens et al. 2009; Arp and Goss 2009) or PFAS may be adsorbed to the filter. If filtration is absolutely necessary, it should be performed in the laboratory, using a validated procedure that includes steps to eliminate the bias that can occur due to sorption issues. As an alternative, laboratory-validated procedures may include centrifuging the sample due to potential filter sorption or contamination issues. See Preparation of Aqueous Samples with Particulates/Suspended Solids in Section 11.2.1.2 for more details. In addition, USEPA Method 1633A strongly discourages composite sampling for Clean Water Act compliance monitoring, stating that samples from sources that flow freely are to be collected as grab samples.
11.1.7.3 Surface Water
Surface water PFAS sampling should be conducted in accordance with traditional methods such as those described in USEPA’s Compendium of Superfund Field Operations Methods (USEPA 1987) with consideration for guidance developed specifically for PFAS by Michigan EGLE (MI EGLE 2024) and New York Department of Environmental Conservation (NY DEC 2023). Depending on the study objectives, sample collection should consider the potential stratification of PFAS in the water body and the tendency for PFAS to accumulate at the air/water interface due to partitioning, which will be of particular concern if foam is observed on the surface (see Section 16.5 on PFAS-containing foam). Transfer containers such as beakers or dippers, which may be attached to extension rods, should be used. For additional information on surface water sampling, see Section 16.4.
- Within the context of sample collection objectives outlined in a site-specific QAPP or work plan, the sample location in the water column should consider the potential stratification of PFAS in solution and their tendency to accumulate at the air/water interface (see Sections 5.2.2.2 and 5.2.4.1). If possible, the sample container or transfer container will be lowered sufficiently below the water surface but above the bottom sediments.
- Transfer containers, such as beakers or dippers, which may be attached to extension rods, should be used only if sample containers have preservatives. Sampling by direct sample container immersion is not recommended in this case.
- Consider using a grab-sampler that seals the sample inside the device at the sample interval and isolates the sample from contact with water above the sample collection interval as the sampler is being removed.
11.1.7.4 Sediment Porewater
Similar in many ways to sampling techniques and equipment used in groundwater sampling for PFAS, porewater purging and sampling involves a variety of materials. The various types of purging and sampling equipment available for porewater sampling are described in Pore Water Sampling Operating Procedure (USEPA 2013). For PFAS sampling, peristaltic pumps with silicon and HDPE tubing are typically used for porewater sample collection, along with push-point samplers, porewater observation devices (PODs), or drive-point piezometers. Push-point samplers and drive-point piezometers are made of stainless steel, while PODs consist of slotted PVC pipe and silicon tubing. PODs and drive-point piezometers are permanent, or dedicated, sampling points typically installed and used for multiple sampling events, whereas push-point samplers are used as a temporary sampling location. Otherwise, the standard procedure for porewater purging and sampling using a peristaltic pump, as described in the Compendium of Superfund Field Operations Methods (USEPA 1987), can be followed. There is information on the development and commercialization of various diffusion-based passive samplers that may be an option to sample porewater within sediment (see Section 11.1.7.6). Installing samplers at interfaces between coarse and fine materials can capture perched porewater (ASTM 2018, section 7.5.2.8). This is consistent with the modeling work by Zeng and Guo (2021) described in Section 10.4.1 in which PFAS was predicted to be found in perched water above capillary barriers.
11.1.7.5 Soil Porewater and Potential Lysimeter Use
The accurate characterization of PFAS in soil porewater within the vadose zone could provide valuable data for understanding the soil-water distribution of PFAS and its impact on groundwater concentration (see Section 10.4.1). One method that has been discussed is the use of lysimeters.
Lysimeters are devices installed within unsaturated soils that sample porewater either actively under positive and/or negative pressure (suction lysimeters) or passively during periods of excess drainage (drainage lysimeters). Lysimeters are widely available commercially and have been used as part of environmental investigations for decades (Singh et al. 2018). Early guidance documents on lysimeter sampling include USEPA (1986) and ASTM (2018).
Sampling of suction lysimeters should occur immediately after infiltration events such as rainstorms, spring melts, or irrigations to capture higher porewater and contaminant flow rates (USEPA 1986, section 4.8.1; ASTM 2018, section 7.5.2.8). Pan lysimeters, or nonsuction porewater sample collection devices, may be more suitable at sites where macropore flow is expected (USEPA 1986, section 4.8.2; and ASTM 2018, section 7.6.1.6).
Prior to installation, lysimeters should be cleaned per the vendor’s standard operating procedure, preassembled, and tested for air leaks. Typically, PVC pipe (equivalent to the depths of the borings) is attached to protect the sample tubes. If potential contaminant interactions with lysimeters is a concern, a laboratory study may be performed before installing at a site (USEPA 1986, section 4.8.3.2).
Like grab samples, lysimeter samples represent a single-time result and should be qualified as such when evaluating impacts to groundwater over a season or from year to year. Although suction lysimeter samples are good for qualitative comparisons, they cannot be used for quantitative PFAS investigation unless the variabilities of parameters involved are established (ASTM 2018, section 7.6.2.1). These parameters may include measured concentrations of PFAS in soil and porewater, as well as pH, permeability, total organic carbon (TOC), and anion and cation exchange capacity (AEC and CEC, respectively) in vadose zone soil; seasonal changes; weather; precipitation; depth; and soil type(s). Also consider measured PFAS concentrations in nearby groundwater wells.
There are limitations to the application of lysimeters, including on sites with:
- shallow groundwater at depths of around 5 feet or less from the surface (consult the manufacturer)
- bedrock that outcrops to the surface
- too little precipitation to result in sufficient soil moisture at depth
- insufficient co-located soil sample data including, but not limited to, TOC, pH, particle size, moisture content, CEC, and AEC
11.1.7.6 Passive Samplers in Aqueous Environments
Each aqueous medium (groundwater, surface water, or wastewater) has specific conditions that affect the transport of PFAS. These conditions will affect decisions regarding the use of passive samplers and the specific device deployed in the field. The following are advantages for the use of passive sampling devices for PFAS in groundwater:
- The elimination or reduction of purge water disposal and other wastes.
- May reduce or eliminate turbidity as compared to pumped samples.
- May reduce both the short-term and long-term costs of sample acquisition.
- Can be used to collect a representative sample in low-yielding wells.
Hydrogeologic conditions required for passive sampling in groundwater (Imbrigiotta and Harte 2020 include:
- Adequate water in the saturated screen interval or the targeted open borehole interval so that the sampling interval is submerged at all times.
- Saturated screens that are not chemically degraded or clogged.
- Adequate residence time for equilibration of hydraulic (well flow), chemical (contaminant concentration), and field physiochemical characteristics (for example, pH, dissolved oxygen).
In most cases with saturated screens/open boreholes 10 feet in length or greater, passive samplers are positioned at the depth of highest mass flux of the constituent of interest. When the interval of highest mass flux is not known, a one-time profiling event can be performed in representative wells using multiple passive samplers deployed at two or more intervals in the saturated screen. When considering transitioning from pumping to passive sampling in wells that have saturated screens 10 feet in length or greater, a favorable comparison of side-by-side results in representative wells suggests that water is being sampled from the same source location by both methods. When the results do not favorably compare, profiling for the interval of highest mass flux is appropriate.
A publication on the use of dual membrane passive diffusion bag (DMPDB) samplersTM for PFAS (Varhol and Varhol 2022) provides results from bench and field tests that show a positive correlation with lab controls and with field samples taken by pumping methods.
Passive sampling methods can be selected to provide time-weighted average (TWA), time-integrated average, or instantaneous temporal data, as well as spatial contamination trends. Passive sampling devices (PSDs) are relatively low cost, simple to use, and do not generate IDW. Some passive samplers can also provide a means of correlating an organismal exposure with a biological effect to determine the biological importance of chemicals sampled by the device (Alvarez et al. 2021; Burki et al. 2006; Vermeirssen, Suter, and Burkhardt-Holm 2006).
There are several different approaches to passive sampling, most of which can broadly be categorized into three types: accumulation, equilibrium, and grab-samplers.
- Accumulation (integrative) devices concentrate the target chemical on a selective collecting medium such as an adsorbent or absorbent solid, a solvent, or a chemical reagent. Target molecules continue to accumulate on the collecting medium during the exposure period and do not come to concentration equilibration with the surrounding medium. The resulting sample mass, or flux, is used to calculate a TWA concentration of target compounds over the exposure period (Huckins, Petty, and Booji 2006; Taylor et al. 2021).
- Equilibrium devices use a semipermeable membrane to contain a collecting medium, which is usually a solvent such as deionized water or uses a performance reference compound (PRC) to calculate a fraction of equilibrium. Target molecules diffuse through the selective membrane and move from higher concentration to lower concentration, in and out of the sampler to maintain a dynamic equilibrium with the surrounding medium. After meeting a minimum residence time, in accordance with the device’s specifications and the DQO’s of the project, the device can be retrieved. Certain samplers can also be left in place at one event and recovered at another. The resulting aqueous sample represents the TWA concentration of target contaminants of the last few days prior to sampler retrieval.
- Grab samplers are devices that acquire a whole media sample in surface water or groundwater, at a specific interval. Once the sample is acquired, the sampler closes to isolate the sample from the surrounding fluids during retrieval. The resulting sample is an aqueous concentration representing the point in time when the sample was collected.
Practitioners have evaluated and used a variety of methods and devices for sampling PFAS in aquatic environments (Taylor et al. 2021; Imbrigiotta and Harte 2020; Alvarez 2010; Gong et al. 2018). A number of PSDs are currently in use and others are being investigated for monitoring PFAS in aquatic environments (Becanova et al. 2021; McDermett et al. 2022; Godlewska, Stepnowski, and Paszkiewicz 2020; Fauvelle et al. 2017; Kaserzon et al. 2012; Wang et al. 2021; Yang et al. 2022; Divine 2020; Varhol and Varhol 2022). Before using any passive sampling device, practitioners should ensure use of the proposed sampler has been validated with respect to the evaluation of the site-specific analytes of interest and is acceptable by the applicable regulatory agency. Refer to ITRC’s Passive Sampling Document for more information on specific devices (ITRC 2024).
11.1.7.7 Sediment
Most core and grab sampling devices are constructed of stainless steel. Some core samplers include an HDPE sleeve inserted in the core barrel to retain the sample. Ensure that materials that contact the media to be sampled do not have water-resistant coatings that contain PFAS. Additional PPE may be required for sampling personnel, such as waders and personal flotation devices. Ensure materials that will potentially contact sampling media do not consist of water-resistant coatings or other PFAS-containing materials or substances. Ensure efficient and consistent homogenization procedures are followed in the field. Refer to Section 11.1.2 for typical materials used during sampling and drilling.
11.1.7.8 Surface Soil
For surface soil sampling, refer to Section 11.1.2 for equipment and supplies, and Section 11.1.5 for decontamination procedures. Ensure efficient and consistent homogenization procedures are followed in the field. No additional considerations are recommended for PFAS sampling of surface soil.
11.1.7.9 Subsurface Soil
Ensure efficient and consistent homogenization procedures are followed in the field. No additional considerations are recommended for PFAS sampling of subsurface soil.
11.1.7.10 Biosolids
Biosolids sampling methods will vary based upon the state and homogeneity of the materials to be collected. Additionally, as the materials are often gathered and stored over days to weeks, varying PFAS concentrations in feedstock can cause varying concentrations within the biosolids matrix unless sufficient mixing is performed.
If high liquid content materials are stored for several hours or days, solids will settle from suspension. Mixing of the storage vessel prior to sampling is essential to ensure collection of representative materials. Dipper, bailer, or pump methods can be used to collect the samples from the midpoint of the storage vessel. If mixing cannot be performed, collection of multiple, stratified, liquid samples followed by compositing may be appropriate.
High solids content materials can often be sampled using traditional compositing techniques. Once again, the accumulation of biosolids over extended periods of time with varying feedstock characteristics can result in potential nonhomogeneous distribution of PFAS within the matrix being sampled. Nonhomogeneity can be reduced through thorough materials mixing and/or use of a high number of grab samples to form a composite for analyses.
For additional information regarding biosolid sampling plans, refer to the NEIWPCC Guide to Biosolids Sampling (NEIWPCC 2006) and the NEBRA PFAS Sampling (NEBRA 2020) guidances.
11.1.7.11 Fish
The Priority Topics for Sampling and Analysis include information about fish sampling and laboratory homogenization (see Section 1.5.2).
The species of fish collected, as well as the portion of fish sampled (whole versus fillet), depends on the project goals (for example, ecological risk or human health). Studies have shown that the majority of the PFAS in fish are stored in the organs, not the flesh (Martin et al. 2004) (Yamada et al. 2014). Communicating project objectives to the laboratory is important prior to fieldwork to determine the necessary quantity and quality of tissue, fish handling requirements, laboratory sample preparation (including single fish or composite fish samples, and whole or fillet preparation), and packing and shipping requirements. According to USEPA Method 1633A, whole fish or other biota samples should be wrapped in aluminum foil or food-grade polyethylene wrap and homogenized tissue samples should be placed in HDPE containers.
11.1.7.12 Air Emissions to Air and Ambient Air
There is an increasing need for the measurement of PFAS in emissions from stationary sources (for example, chemical manufacturing, industrial use, combustion and thermal treatment and other treatment systems not originally designed to treat PFAS from exhaust), as well as in ambient air. Due to the diverse nature of PFAS, multiple measurement approaches are needed to measure polar and nonpolar, volatile, semivolatile, and nonvolatile (particulate-bound) PFAS.
Emissions to Air
As of the date of this publication, there are no multilaboratory-validated sampling methods for PFAS in air emissions (for example, from thermal treatment in manufacturing plants or incinerators). In their absence, emissions measurements have been performed using modifications to USEPA SW846 Method 0010 (Modified EPA Method 5 Sampling Train) (USEPA 1986), a method designed for measurement of semivolatile organic compounds. Other methods have been adapted to capture specific individual compounds of interest.
USEPA and European groups (Verein Deutscher Ingenieure [VDI], an association of German engineers) are evaluating and investigating which sampling and analytical methods might be, in principle, the most suitable to capture PFAS and resulting byproducts in all fractions of the emissions (particles, moisture, gas phase).
PFAS can be partitioned in stack emissions into several different fractions due to the physical properties of these species. At the elevated temperatures typically encountered in stack emissions the vapor pressure can be sufficiently high that some PFAS are present in the gas phase. The lower molecular weight fluorotelomer alcohols (FTOHs) have lower boiling points and so may primarily be present as vapors. PFAS can adsorb to particulate matter, are highly water soluble, and can dissolve in water droplets if present in the stack. To measure these partitioned fractions, the stack effluent is sampled isokinetically (that is, the air enters the probe at the same velocity as it is moving in the stack, to accurately sample particles and droplets) and captured on a heated filter, an XAD-2 (polymer of styrene divinyl benzene) sorbent resin tube, and in water impingers. In some test programs a second XAD-2 sorbent cartridge is included in the sample train to determine if breakthrough has occurred. The filter, sorbent cartridge, and water impingers are recovered separately, and the sample train components are rinsed with a methanol/ammonium hydroxide solution.
In 2021, USEPA released Other Test Method (OTM) 45 Measurement of Selected Per- and Polyfluorinated Alkyl Substances from Stationary Sources (USEPA 2021). In July 2024, USEPA released Revision 1 to OTM-45 (USEPA 2024). This performance-based method was made available by USEPA as a recommended method that can be used to measure 50 specific semivolatile and nonvolatile polar PFAS from a variety of stationary sources. USEPA OTM-45 is largely based on the USEPA SW-846 Method 0010 (Modified EPA Method 5 Sampling Train) with several modifications. PFAS are collected in four sample fractions: 1) filter; 2) primary XAD-2; 3) impingers (containing water); 4) secondary XAD-2 (for breakthrough determination). Each fraction, with its associated rinses, is extracted and analyzed separately. Analyses are performed by isotope dilution LC/MS/MS. The analytical approach is based largely on EPA Method 533 and includes many of the same analytical qualification criteria, with the additional requirement of monitoring confirmatory secondary transition ions, similar to USEPA Method 1633A. USEPA OTM-45 was released as an “Other Test Method (OTM)” by USEPA’s Emission Measurements Center to promote consistency and is considered by USEPA to represent the current best practices to sample and analyze PFAS from stationary sources. This method is under evaluation and will be updated as more data from stakeholders become available. Field sampling programs must include collection of field blanks as a means of assessing PFAS artifacts present in sampling media and potentially introduced during sample handling in the field. Other QC measures that should be considered include the use of isotopically labeled PFAS field spikes, which are useful for quantifying losses associated with field collection techniques. For a PFAS field spike, isotopically labeled spike compounds are typically applied/spiked by the laboratory into the XAD-2 sorbent media prior to field deployment. These compounds serve to assess analyte (“native PFAS in air”) collection efficiency, breakthrough, and the accuracy of the combined sample collection and analysis method on a sample-specific basis.
In 2024, USEPA released OTM 50 Sampling and Analysis of Volatile Fluorinated Compounds from Stationary Sources Using Passivated Stainless-Steel Canisters (USEPA 2024). This performance-based method was made available by USEPA as a recommended method that can be used to measure a list of 30 specific volatile fluorinated compounds (VFCs) indicative of incomplete decomposition of some PFAS from a variety of stationary thermal treatment sources. This method also allows for the detection of additional VFCs and non-fluorinated volatile compounds via non-targeted analysis. Samples are collected using a 6-liter passivated silicon ceramic lined stainless-steel canister or equivalent. Samples containing low moisture (<3% volume/volume) or acid content can be sampled directly into the canister; otherwise, a moisture and acid management system comprised of a filter and impingers system similar to that which is included in OTM-45 is utilized. Like OTM-45, this method is under evaluation and will be updated as more data from stakeholders become available.
USEPA ORD is researching and evaluating multiple PFAS measurement approaches for polar and nonpolar, volatile, semivolatile and nonvolatile PFAS, including further development of USEPA OTM-45 for nonpolar PFAS. The development of an additional method, OTM-55, is currently underway by USEPA to measure nonpolar semivolatile and volatile PFAS, including fluorotelomer alcohols (FTOHs) and products of destruction (PIDs) using whole canister sampling and analysis. USEPA ORD is also evaluating the use of Fourier Transform Infrared (FTIR) spectroscopy to measure select volatile PFAS in real-time. Several of the PFAS capable of being measured are also being evaluated as potential indicators or surrogates of PFAS destruction performance.
Multiple PFAS emissions tests have been performed at a variety of sources for multiple purposes including source characterization, assessment of control technology performance, and evaluation of treatment technologies. Stationary source, or stack, emissions of PFAS have been measured in North Carolina (NC DEQ 2019) and New Hampshire (NH DES 2019) from industrial facilities that synthesized PFAS (Chemours, NC) or used PFAS in manufacturing processes (Saint Gobain Performance Plastics, NH) (Beahm and Marts 2019). These test programs confirmed that stack emissions from industrial facilities contribute to ground and surface water contamination (NC DEQ 2019). An additional study at Chemours, NC, reported on the commissioning of an installed thermal oxidizer control system (Weston Solutions, Inc. 2020).
Ambient Air
There are no USEPA Federal Reference Methods (FRM) or Toxic Organic Methods (TO series) available specifically for the measurement of PFAS in ambient air. In their absence, some sampling and analysis of ambient air have been performed using modified TO methods, such as TO-13A and TO-9 (USEPA 2020). Both methods use high-volume air samplers fitted with a particulate glass fiber filter/quartz fiber filter (GFF/QFF) and sorbent cartridge for the collection of particulate and gaseous phases, respectively. USEPA TO-13A specifies collection of air samples at a flow rate of approximately 225 liters/minute, resulting in an air volume greater than 300 m3. The solid sorbent used consists of a “sandwich” of polyurethane foam (PUF) and XAD-2.
PFAS in ambient air have been measured using both active (with actual flow) and passive (gas diffusion) sampling techniques. Most techniques have made use of solid sorbents such as PUF, XAD-2, and sorbent-impregnated PUF (SIP). (Finely ground XAD-4 resin is often the sorbent of choice for impregnating the PUF). Active samples also include a particulate filter (glass or quartz fiber) ahead of the sorbent module. To optimize detection limits, high-volume air samples have been used most often.
Detection limits of air and emissions methods can be greatly influenced by PFAS artifacts found in the neat filter, sorbent media, or components within the sampler itself. For example, use of Teflon gaskets in high-volume samplers is not recommended. Field sampling programs must include collection of field blanks as a means of assessing PFAS artifacts present in sampling media and potentially introduced during sample handling in the field. Other QC measures that should be considered include collection of field duplicate or co-located samples and the use of isotopically labeled PFAS field spikes. The latter compounds are typically applied/spiked by the laboratory into the sorbent media prior to field deployment. These compounds serve to assess analyte (“native PFAS in air”) collection efficiency, breakthrough, and the accuracy of the combined sample collection and analysis method on a sample-specific basis.
Passive samplers should also make use of mass-labeled PFAS as a sample-specific quality control measure to account for native PFAS losses during each sampling event. Volatilization of labeled PFAS during the deployment period provides sampling rates on a site-specific basis and accounts for both temperature and wind influences.
USEPA and European groups (VDI) are evaluating and investigating which sampling methods might be, in principle, the most suitable to capture PFAS and resulting byproducts in all fractions of the emissions (particles, moisture, gas phase). An important consideration is that fluorinated polymers are used in common sampling equipment, which may cause contamination of the samples. For the purposes of PFAS determinations, this material must be replaced. In addition to concerns over using fluorinated polymers in sampling equipment being a source of contamination, there are also concerns about the potential for sorption of PFAS to fluorinated polymers, thereby effectively reducing the observed concentration, or affecting any attempt to quantify the phase distributions between condensed and non-condensed phases (for example. PM vs gaseous). This is related, in part, to the concerns about quantification of ambient air concentrations and distributions, as published by Johansson, Berger, and Cousins (2017), showing that the use of GFF (and chemically deactivated glass fiber filters) may irreversibly bind fluoro-carboxylates during collection of samples containing fluoro-acids, which can be in the gas phase depending on their vapor pressure and ambient temperature, and the corresponding carboxylates. This issue for ambient air appears to negatively impact the estimates of phase distributions and so far, may not have a technique that is applicable for quantitative recovery (no answer to this problem has yet been published). A related issue, though separate from the ambient sampling confounding issues, is that quantification of acids/carboxylates via LC/MS does not/cannot distinguish between these two oxidation states, which is important to the phase distribution in ambient air (and emissions to air). There are other techniques (GC/MS or Chemical Ionization Mass Spectrometry (CIMS) (Riedel et al. 2019) that may help address these LC/MS deficiencies, particularly with regard to air measurements.
11.1.7.13 Human Blood, Serum, Tissue
There are no official or standardized methods for testing human blood, serum, or tissue. Laboratories and the Centers for Disease Control and Prevention (CDC) are in the process of developing best methods. A procedure developed by the CDC’s National Center for Environmental Health has been published (CDC 2016). There are also several laboratories advertising this capability; however, the analytical methods and modifications from validated environmental laboratory protocols may not be consistent among these vendors. Human testing is outside the scope of this document; however reference points that could be used for comparison of whole blood or serum results to geometric mean serum levels generated from the U.S. population are included in the ATSDR ToxGuide for Perfluoroalkyls (ATSDR 2020).
11.1.7.14 Potential High Concentration Samples
The CSM or historical data may indicate areas of high concentrations of PFAS for which single-use, disposable equipment is recommended. If single-use is not possible, take additional precautions such as implementing a greater frequency of ERBs and not reusing equipment to sample potentially low PFAS concentration samples. High concentration samples may require smaller sample volumes to be collected for analysis. High concentration samples should be segregated during shipping to the laboratory, and clearly identified on the sample chain-of-custody form.
Some projects may require the analysis of AFFF product that has been used at the site. All AFFF product samples must be considered high concentration samples. The method DOD AFFF01 contains steps to prepare AFFF concentrate samples. This method requires that a dilution of a subsample of the AFFF concentrate be prepared for extraction. A critical step in this procedure is the amount of time that must elapse, a minimum of 3 hours (Willey 2021) from the time PFAS-free reagent water is added to the aliquot of AFFF concentrate to create the dilution to the time when extracted internal standard can be added to the diluted sample. It was determined during method development and validation that some AFFF concentrates can take up to 3 hours to fully dissolve in the reagent water. In addition, this method requires each AFFF concentrate sample to be prepared and analyzed in duplicate, using another aliquot of the collected sample. It is recommended that these samples be segregated from other samples during sampling and shipping to avoid cross-contamination and clearly identified on the sample chain-of-custody form.
11.2 Analytical Methods/Techniques
11.2.1 Quantitative Techniques
11.2.1.1 General
Analytical methods are still evolving for PFAS analysis. Several methods are multi-laboratory validated and published. Three multilaboratory-validated methods, USEPA Methods 537, Version 1.1 (herein referred to as USEPA 537), 537.1, and 533 have been published for analysis of drinking water samples (USEPA 2009; USEPA 2020; USEPA 2019) and two methods for non-drinking water matrices have been published. USEPA Method 1633A (USEPA 2024) has been published for analysis of groundwater, surface water, waste water, soil, sediment, biosolids, tissue, and landfill leachate. USEPA SW-846 Method 8327, has been published for analysis of groundwater, surface water, and wastewater samples. The DOD has published a multilaboratory validated method, DOD AFFF01 (Willey 2021), for the analysis of AFFF concentrates to demonstrate compliance to the military specification for AFFF (MIL-PRF-24385). The USEPA released OTM 45 (USEPA 2021) and OTM 50 (USEPA 2024) for measurement of PFAS in emissions to air from stationary sources. Also in January 2024, USEPA released draft OTM 50 for measurement of fluorinated compounds in emissions to air from stationary sources by GC-MS.
- USEPA Method 537, Version 1.1 tests for 14 PFAS analytes
- USEPA Method 537.1 tests for 18 PFAS analytes (including 4 PFAS not included in USEPA Method 533)
- USEPA Method 533 tests for 25 PFAS analytes (including 11 not included in USEPA Method 537.1)
- USEPA SW-846 Method 8327 tests for 24 PFAS analytes (does not include all PFAS included in USEPA Method 537.1 or 533)
- USEPA Method 1633A tests 40 PFAS analytes (includes all PFAS included in USEPA Method 537.1, 533, and 8327 and 11 additional PFAS analytes)
- DOD AFFF01 tests for PFOA and PFOS
- USEPA Other Test Method (OTM) 45 tests for 50 specific semivolatile and nonvolatile polar PFAS
- ASTM D8421-22 Standard Method for Determination of Per- and Polyfluoroalkyl Substances (PFAS) in Aqueous Matrices by Co-solvation followed by Liquid Chromatography Tandem Mass Spectrometry (LC/MS/MS)
- Draft USEPA OTM 50 tests for 30 specific volatile nonpolar fluorinated compounds
Other methods have been published by other organizations. Lists of these methods, by various categories, are provided in the PFAS Analytical Methods Excel File in the separate tabs:
- External Table 11-2–Published Method Basics. Provides information on basic principles of each method (media type, validation status, method type, sample container requirements, holding time, preservation requirements, and analytical instrument.
- External Table 11-3–Published Methods Specifics. Provides more details of the methods such as sample preparation requirements, quantitation scheme, confirmation ion requirements, quantitation limits, and isomer profile.
- External Table 11-4–Provides summaries of the analyte lists for each of the methods. The method analyte list for each of these methods varies.
- External Table 11-5–Draft Published PFAS-Related Analytical Methods Basics. Provides information on basic principles of each method (media type, validation status, method type, sample container requirements, holding time, preservation requirements, and analytical instrument).
11.2.1.2 Sample Preparation
USEPA Methods 537, 537.1, 533, and 1633A, and DOD AFFF01 all require aqueous samples to be prepared using the solid-phase extraction (SPE) technique, but may use different solid-phase selectivities. USEPA Method 1633A and DOD AFFF01 both require cleanup procedures be used on extracts and all associated batch QC samples to help eliminate matrix interferences (for example, bile salts, gasoline range organics) that could be present. USEPA Methods 537.1 and 533, USEPA Method 1633A, and DOD AFFF01 all require extraction of the entire sample collected (in the case of DOD AFFF01, the entire prepared dilution), including a rinse of the sample container (in the case of DOD AFFF01, the dilution container).
USEPA SW-846 Method 3512 is for the preparation of groundwater, surface water, and wastewater samples by diluting the sample collected with an equal volume of methanol and does not require a rinse of the sample container to be included unless the sample had to be transferred to a larger container in order to allow for the addition of the appropriate volume of methanol. USEPA SW-846 Method 3512 does not use SPE or carbon clean-up steps, which is another significant difference from the other USEPA published methods. USEPA SW-846 Method 8327 states that during method development, some PFAS analytes showed a potential for loss during sample preparation or standard preparation using USEPA SW-846 Method 3512. This method states that a minimum organic cosolvent content must be maintained in standards and samples, and it cautions against aqueous subsampling prior to adding sufficient organic solvent.
USEPA Method 1633A prepares groundwater, wastewater, surface water, and landfill leachates using solid phase extraction (SPE) followed by carbon clean-up steps to eliminate matrix interferences. USEPA Method 1633A uses solvent extraction for soil, sediment, biosolids, and tissue preparation, followed by use of SPE cartridges and carbon clean-up steps to eliminate matrix interferences. Regardless of the method used, care must be taken to prevent sample contamination during preparation and extraction because the limits of quantitation and detection are 1,000 times below (ppt) those for more routine analyses such as volatiles or semivolatile analysis (ppb). It is recommended that all supplies be checked and confirmed as PFAS-free prior to sample preparation. Intermittent contamination can occur due to vendor or manufacturing changes.
Some PFAS analytical methods, such as USEPA Method 533 and USEPA Method 1633A, use isotope dilution and extracted internal standard quantification schemes to calculate sample concentrations. Isotope dilution analysis (IDA) quantitation requires the use of extracted internal standards (EIS) that are the isotopically labeled analogs of the method analytes. Method analytes for which corresponding isotopically labeled analogs are not commercially available are quantitated using the isotopically labeled analogs of a method analyte of similar chemical properties. Since isotopically labeled analogs of PFOA and PFOS are commercially available, DOD AFFF01 uses isotope dilution quantitation. USEPA SW-846 Method 8327 uses an external quantitation scheme to calculate the percent recovery of isotopically labeled analogs that are added to the sample prior to dilution with methanol. This method does not use isotopically labeled analog recoveries to account for sample preparation and matrix interference biases in the sample result. All of the methods discussed above require these isotopically labeled standards be added to the sample at a designated point in sample preparation, depending on the sample matrix:
- aqueous samples–added to field samples while in the original container prior to extraction/dilution
- AFFF concentrates or high concentration aqueous samples – added to dilution of sample prior to extraction
- solid samples and biota–added after homogenization and subsampling, prior to addition of water or extraction solvent
Ensuring a representative sample/subsample for analysis is critical. For aqueous samples, the entire sample and rinsate of the sample container received by the laboratory must be extracted by SPE to recover any PFAS that adhere to the sample container. Filtration is not recommended for samples with high particulate content because retention of PFAS onto filters has been noted. Centrifugation is often used to reduce or separate out sample particulates. See Preparation of Aqueous Samples with Particulates/Suspended Solids below for more details.
USEPA Method 1633A requires the screening of all aqueous samples using a separate sample container from the one which will be used for sample preparation, absent project-specific requirements. Due to limitations in SPE cartridge capacity and potential contamination of sample preparation and/or analytical equipment, the method requires samples containing high concentrations of PFAS (for example, AFFF concentrates) to be diluted prior to SPE and sample clean-up. In these cases, sorption onto the original sample container is not an issue, depending on the identified project-specific DQOs, because the amount of PFAS adsorbed onto the container walls may be negligible compared to the amount of PFAS in the sample.
USEPA Method 1633A requires the entire soil, sediment, and biosolid sample that is collected be homogenized in the laboratory prior to subsampling. Tissue samples are to be prepared according to project requirements (for example whole fish versus fillet) and homogenized prior to subsampling.
Cleanup procedures can be used on extracts and all associated batch QC samples (for example, method blank and laboratory control samples) when matrix interferences (for example, bile salts, gasoline range organics) could be present. USEPA Method 1633A and DOD AFFF01 require carbon cleanup techniques to be used for all sample matrices. USEPA Method 1633A and DOD AFFF01 require samples to be slowly concentrated to remove methanol from the extract to avoid loss of neutral and other highly volatile method analytes. In addition, USEPA Method 1633A states that if methanol is not sufficiently removed, its presence during SPE can result in poor recovery of long-chain carboxylic acids and sulfonates. Care must be taken to avoid these outcomes.
The preparation batch QC samples that are required by these PFAS methods vary. Common laboratory QC sample nomenclature and typical use frequencies are as follows:
- Method blank – (one per preparation batch of 20 or fewer field samples) PFAS are ubiquitous and found in many analytical instrument systems, reagents, containers, and common laboratory environments. The method blank is a similar matrix as associated field samples and undergoes the same sample preparation procedure as the associated field samples. It is a vital indicator of the absence of PFAS contamination in laboratory equipment, supplies, and reagents. Note that method blank is also referred to as Laboratory Reagent Blank in EPA drinking water methods.
- Sample (or laboratory) duplicate – (minimum of one per preparation batch of 20 or fewer field samples) A sample that is prepared and analyzed in duplicate in a single laboratory to ensure the laboratory’s subsampling procedures are capable of achieving a known level of precision as defined in the QAPP or work plan.
- Laboratory control sample (LCS), ongoing precision and recovery (OPR), or laboratory fortified blank (LFB) – (typically one per preparation batch of 20 field samples) Must contain all project-specific PFAS in similar media as the associated field samples and is used to evaluate bias associated with sample preparation as well as analytical processes.
- Low-level laboratory control sample (LLLCS) or low-level ongoing precision and recovery (LLOPR) – (one per preparation batch of 20 field samples) Must contain all project-specific PFAS at a specified concentration (for example, 0.5–2 times the LOQ for USEPA SW-846 Method 8327 or 2 times the LOQ for USEPA Method 1633A) in similar media as the associated field samples and is used to evaluate bias associated with sample preparation as well as analytical processes.
- Certified reference material (CRM) – (if available, one per preparation batch of 20 or fewer field samples) Unlike LCSs, which contain no matrix interferences, CRMs can be of significant value when dealing with complex matrices such as soil, sediment, and tissue.
- Matrix spike (MS) and MS duplicate (MSD) – (one pair per prep batch of 20 or fewer field samples) An MS/MSD is not needed if IDA can be used for all PFAS of interest because the EIS recoveries account for the influence of matrix interferences in each sample. If EIS standards are not available for a PFAS of interest, an MS/MSD may be warranted to assess the effects of matrix interference on that specific PFAS.
For samples with high concentrations of PFAS, it is recommended that an LCS duplicate and a sample duplicate are prepared in lieu of an MS and MSD. DOD AFFF01 requires the sample duplicate to be prepared using a different aliquot from the same sample bottle to create the sample duplicate.
Preparation of Aqueous Samples with Particulates/Suspended Solids
USEPA Method 1633A contains a procedure for determining the solids content of aqueous samples. This determination is to be performed using the second, smaller container that is required for each sample, not the sample container used to prepare the sample for the PFAS analysis. The sample preparation procedures for PFAS analysis in USEPA Method 1633A are applicable to aqueous samples containing less than 50 mg of solids. The method provides additional steps to take if particulates clog the SPE cartridge during extraction, including the use of additional SPE cartridges. Solids accumulate high concentrations of PFAS and specifically some of the longer-chain PFAS. Care should be taken to resuspend any solids and to rinse bottles so that measured concentrations reflect the entire sample.
When aqueous samples contain greater than or equal to 50 mg of solids, depending on the laboratory procedure, if the contaminant mass in the solids is not determined, the reported PFAS (fingerprint) could be inaccurate for effective comparison. Ultimately, the data user needs to work with the laboratory to determine the appropriate procedure to be performed, depending on the end use of the data (for example, remedial action, human health or ecological risk assessment, permit compliance, etc.). The project objectives may specify quantification of the total aqueous sample, water plus solids, or may only include evaluation of the aqueous fraction for end use as drinking water, where the solids will not be consumed.
A survey conducted in 2020 by ITRC received responses from 16 laboratories. These responses demonstrated that the laboratory community is not consistently preparing aqueous samples in the presence of suspended solids.
- Several laboratories centrifuge the samples and decant off the water portion for subsequent extraction.
- Some laboratories may also extract the remaining particulates and combine the extract with the aqueous extract. However, only one of the surveyed laboratories does this routinely; the others do this only if requested by the client.
- Routine laboratory procedures are not dependent upon whether the matrix is groundwater, surface water, or wastewater.
- Several laboratories filter the samples, if requested by the client. This may result in sorption of PFAS from the samples and potential cross-contamination, and should be implemented with care.
- After decanting, some laboratories rinse the remaining particulates in the bottle with solvent for the SPE.
- Not all laboratories disclose when samples require special preparation (decanting, centrifuging, or filtering) due to the presence of particulates. Therefore, this would not always be known to the data user.
- Laboratories have different procedures for when particulates clog the SPE cartridge; some labs may re-extract at a dilution, while others may start a new cartridge and attempt to continue with the extraction of the remaining sample.
Laboratories should clearly state whether reported concentrations are dissolved (water only) or “total” PFAS measurements (including sample particulates). The project team will determine whether dissolved or “total” PFAS measurements are appropriate to meet project objectives and confirm with laboratories that correct procedures are in place. Sampling procedures should be designed to gather representative samples to meet project objectives. Some issues to consider in the determination of the need for a total or dissolved PFAS measurement are discussed in this section.
Groundwater
- If minimizing presence of particulates is within the project objectives, use of low-flow groundwater sampling or no-purge samplers should be considered.
- If turbidity is >10 NTU, consider a “total” measurement if sampling groundwater for compliance, delineation, remedial design, or risk assessment. “Total” can be defined as centrifuge, decant, and extract both phases, to report the dissolved concentration and the suspended/solid concentrations either individually or summed in the report.
- Collect samples for TSS analysis to assist in the evaluation of sample data. In lieu of TSS, turbidity measurements can also be helpful in the evaluation.
Drinking Water
- Particulates are not addressed in the drinking water methods.
Surface Water and Wastewater
- Use a “total” measurement if sampling surface water or wastewater for compliance, permitting, remedial design, or risk assessment.
- A “total” measurement may not be required if sampling for a line of evidence in a source area, rather than for a regulation.
- Collect samples for TSS analysis to assist in the evaluation of sample data. In lieu of TSS, turbidity measurements can also be helpful in the evaluation.
For laboratories performing centrifugation and subsequent decanting, it should be noted that it is important that the extracted internal standards are spiked into the aqueous samples prior to centrifugation. It is important to communicate with your laboratory prior to the collection of samples on the approach that will be used.
Biosolids
Biosolids sample preparation methods can vary based on laboratory-specific protocols, the moisture content of the sample, and/or client or regulatory requirements. Because of the variability in sample preparation methods for this matrix, communication with the laboratory prior to submitting samples is essential to understand the laboratory’s options for sample handling and to ensure that the laboratory will meet the project objectives and/or regulatory requirements.
The following preparation methods are possible for biosolids and may be dependent on the laboratory’s standard operating procedures.
- Typical solid sample extraction procedures (e.g., homogenization and solvent extraction) may be used regardless of the moisture content if there are visible or settleable solids. Because regulatory criteria for biosolids are typically established on a mass/unit mass basis (e.g., µg/kg), this may be an approach needed to satisfy regulatory requirements.
- If there are no visible or settleable solids, the laboratory may treat the biosolids sample as an aqueous sample (e.g., passing the sample through a solid phase extraction [SPE] cartridge).
- When aqueous samples are treated as dissolved PFAS samples, laboratories may centrifuge the samples to separate out any solids or particulates. The aqueous phase would then be decanted and prepared using SPE.
- Some laboratories may also extract the remaining solid/particulate phase after decanting using a solvent and then use the eluate of this solid sample to elute the PFAS off the SPE cartridge used to extract the aqueous phase, thus combining the aqueous and solid phases into one extract.
- If samples are not centrifuged, solid particles may become entrained on the SPE cartridge and PFAS from these solid particles may be extracted during the methanol rinse. However, there is also a chance that the solid particles could cause clogging of the SPE cartridge, which would not allow for a complete extraction.
Laboratories may also be able to perform biphasic extraction and report results separately for the aqueous and solid phases.
11.2.1.3 Sample Analysis
Instrument Type–LC/MS/MS
All methods published by the USEPA and DOD for targeted PFAS analysis use liquid chromatography-mass spectrometry-mass spectrometry (LC/MS/MS), which is especially suited for analysis of ionic compounds, such as the PFSAs and PFCAs. Gas chromatography-mass spectrometry (GC/MS) can also be used for PFAS analysis, specifically the neutral and nonionic analytes, such as the fluorotelomer alcohols (FTOHs), perfluoroalkane sulfonamides, and perfluoroalkane sulfonamido ethanols. GC/MS may be appropriate for (ambient) atmospheric samples. PFAS are either directly detected using large-volume injection (Scott et al. 2006) or detected as a derivatized compound that is GC/MS amenable (Langlois et al. 2007). At this time, OTM 50 is the only published GC/MS method and there is very limited commercial availability for this technique for PFAS analysis.
In contrast, LC/MS/MS analysis of PFAS is widely available. LC/MS/MS is typically operated in multiple reaction monitoring (MRM) mode, which allows for selective monitoring of both the precursor and (potentially) unique daughter ions that are produced upon fragmentation in the mass spectrometer; this allows for specific and selective monitoring for compounds of interest. MRM is performed by specifying the mass-to-charge ratio of the compound of interest for fragmentation within the MS/MS. The precursor mass of the compound of interest undergoes MS/MS fragmentation, followed by monitoring for product ions. Ions arising from that fragmentation are monitored for by the MS/MS, which yields improved specificity and sensitivity.
Standards Preparation and Storage
Certified analytical standards are available from several manufacturers. Products may have variable purity and isomer profiles, which may compromise the accuracy, precision, and reproducibility of data. Certified standards of the highest purity available are standards that can be used for accurate quantitation. Standards manufacturers should provide a certificate of analysis (COA) with each standard, which provides the analyte concentrations and the purity information. Regarding expiration dates, laboratories should have their own policy in place for expiration date determination.
Quantitative standards containing linear and branched isomers are not commercially available for all applicable analytes. As of the date of this publication, quantitative standards are available for PFOA, PFOS, perfluorononanoic acid (PFNA), perfluorohexane sulfonic acid (PFHxS), perfluorooctane sulfonamide (PFOSA), N-methyl perfluorooctane sulfonamide (NMeFOSA), N-ethyl perfluorooctane sulfonamide (NEtFOSA), 2-(N-methylperfluorooctanesulfonamido) acetic acid (NMeFOSAA), 2-(N-ethylperfluorooctanesulfonamido) acetic acid (NEtFOSAA), methyl perfluorooctane sulfonamido ethanol (NMeFOSE), and N-ethyl perfluorooctane sulfonamido ethanol (NEtFOSE).
Technical grade standards, or qualitative standards, that contain branched and linear isomers are available for other PFAS, but these standards do not have the accuracy needed for quantitation purposes. USEPA Method 1633A and DOD AFFF01 both require the analysis of technical grade standards to verify which peaks represent the branched isomers for method analytes when these standards are available.
Stock standards of PFAS analytes, internal standards, and surrogate standards supplied in glass ampoules by the manufacturer are acceptable. Manufacturers of the certified analytical standards often provide laboratories storage and shelf-life guidance for stock and working standards. USEPA Methods 533, 537.1, 8327, Method 1633A, OTM-45, and DOD AFFF01 contain storage requirements for both reagents and/or standards.
Steps to Help Eliminate Laboratory/Instrument Contamination (Verification of Supplies, Instrument Blanks, Isolator/Delay Columns)
Laboratory and instrument contamination is of particular concern for PFAS, given that the limits of detection are in the ppt range. Additionally, nonpolymer PFAS may be found at trace levels as impurities in some polymer products (3M 1999). PFAS are found in commonly used laboratory items such as PTFE products, solvent lines, and methanol, which could lead to method interferences and elevated baselines in chromatograms if not checked. The evaluation criteria for equipment and standards that is applicable depends on the analytical method used. For instance, USEPA Methods 537.1 and 533 recommend that all of the above items must be “less than 1/3 the MRL (minimum reporting limit) for each method analyte under the conditions of analysis by analyzing laboratory reagent blanks.” (USEPA 2020, pg. 7). USEPA Method 533 further specifies that the isotopically labeled analogs of method analytes and isotope performance standards meet this same requirement (USEPA 2019, pg. 7).
Most vendors have a PFAS kit available that replaces as many PFAS-containing components as possible. In addition, the liquid chromatograph can be fitted with an isolator or delay column to separate contamination arising from the solvent delivery systems. This can reduce the contribution of PFAS from the pump apparatus to the analysis and reduce detection limits. Guard columns should be used to protect analytical columns.
Ion Transition Selection (Recommended Transitions for Primary and Confirmation Ions, Including Ratio Criteria)
Quantification by LC/MS/MS may be accomplished using a variety of techniques. For relatively simple matrices such as drinking water, USEPA Method 537.1 quantifies analytes by comparing the product ion of one precursor ion and retention time in samples to calibration standards. For more complex matrices, additional product ions and their ion ratios can be used to distinguish analytes from matrix interference. In an MS/MS system, most analytes can be fractured into more than one ion. By monitoring the area of each ion and comparing the ratio of those area counts, a more definitive identification can be made. This identification allows the analyst to distinguish true target analytes from false positives. This more detailed quantification is not required for drinking water matrices, but it is useful for more complex matrices. USEPA SW-846 Method 8327, USEPA Method 1633A, and DOD AFFF01 all require two ion transitions from precursor to characteristic product ions be monitored and documented for each analyte, with the exception of those analytes without a suitable secondary transition (for example, species with nondetectable or low signal secondary transitions, such as PFBA, PFPeA, PFMPA, and PFMBA). These methods determine ion transition ratio criteria by comparison with the analysis of analytical standards and use these ratios to evaluate potential bias in sample results.
Mass Calibration, Calibration Criteria, and Calibration Verification
All LC/MS/MS instruments require mass calibration prior to initial calibration. Mass calibration and mass calibration verification should be performed at setup, after performing maintenance that is required to maintain instrument sensitivity and stability performance, and as needed based on QC indicators, such as calibration verifications, as required by USEPA Method 1633A. Mass calibration should be performed according to the manufacturer’s instructions. In addition to the manufacturer-specified mass calibration verification, the method also requires the accuracy of the ion masses monitored by the method be verified.
Following mass calibration and mass calibration verification, an initial calibration should be performed and verified. All of the published USEPA methods and the DOD method contain requirements for instrument calibration and calibration verification specific to the PFAS concentrations expected in the media for which the method is applicable. USEPA Method 537.1 uses internal standard quantitation, while USEPA Method 533 and USEPA Method 1633A use isotope dilution and extracted internal standard quantitation. USEPA SW-846 Method 8327 uses external calibration quantitation. Isotope dilution quantitation is recommended for complex matrices. The instrument is required to be calibrated at setup and as needed following QC failures, such as initial calibration verification (ICV) or continuing calibration verification (CCV) failure. The lowest calibration point should be a concentration at or below the Minimum Reporting Limit (MRL), Minimum Level (ML), or Limit of Quantitation (LOQ), depending on the method. Most methods require the analysis of a standard at the MRL, ML, or LOQ concentration at least daily to document the instrument’s ability to accurately quantitate down to that concentration. In addition, some methods also require a reporting limit check QC sample (for example, LLLCS or LLOQ verification sample) to be included with each sample preparation batch to demonstrate adequate quantitation at the lowest concentration is achievable using the sample preparation techniques required by the method.
Some methods, such as USEPA SW-846 Method 8327, require analysis of an ICV, which is a calibration verification standard prepared from a source separate from the calibration standards and analyzed after each initial calibration and before sample analyses are performed.. CCVs or CVs should be analyzed at the frequency specified in the analytical method. Most USEPA methods require at a minimum a CCV/CV to be analyzed prior to sample analysis on days an ICV is not analyzed, after every 10 field samples, and at the end of the analytical sequence. Some methods rotate the concentration of CCVs/CVs to cover the entire calibrated range of the instrument and vary the acceptance criteria depending on the concentration. For example, in USEPA Method 537.1, the calibration acceptance criteria for each analyte at the lowest calibration point must be within 50–150% of its true value while the other calibration points must be within 70–130% of the true values.
Isotope dilution and extracted internal standard quantitation can correct bias resulting from loss during sample preparation, such as in USEPA Methods 533 and 1633A. In the case of USEPA SW-846 Method 8327, isotopically labeled analogs are used as surrogates to monitor for loss without being used for recovery-correction of target analyte concentrations. Isotope dilution is a quantitation technique that considers sample matrix effects on each individual PFAS quantitation in the most accurate and precise manner possible. This technique quantifies an analyte of interest against the isotopically labeled analog of that analyte, which is added to the sample prior to the onset of sample preparation. EIS quantitation is the comparable technique used when an isotopically labeled analog of an analyte is not commercially available. Addition of EIS prior to sample preparation helps account for loss of analyte during the preparation process and for bias associated with the instrumentation. Calibration criteria for methods using isotope dilution and EIS quantification schemes can be found in USEPA Methods 533 and 1633A. Methods using isotope dilution should include isotopically labeled analog recovery for each sample and analyte in data reports. Isotopically labeled analog recoveries should be reported, and minimum/maximum isotopically labeled analog recoveries may be required by specific analytical procedures. For instance, USEPA Method 1633A determines the recovery of these analogs through comparison to the response of analogs typically called non-extracted internal standards (NIS) added to the sample after extraction and prior to analysis. Depending on project DQOs, low isotope recovery may indicate that quantitation was inadequate; the data are then reported as estimated values or not at all.
Instrument Blanks: When Are They Needed, Criteria, and Corrective Actions to Take
Due to the extensive use of PFAS, instrument blanks are critical in determining if the instrument is potentially affecting PFAS concentrations in samples. Some methods, such as USEPA Method 1633A, require instrument blanks to be analyzed following the highest calibration standard analyzed, daily prior to sample analysis, after each CCV/CV, and following samples that exceed the calibration range to ensure carryover does not occur. The acceptance criteria for instrument blanks are dependent on the method. For example, USEPA SW-846 Method 8327 requires the concentration of each analyte to be ≤ ½ the LLOQ or < 10% of the sample concentration. USEPA Method 1633A is the only method which states that if instrument blanks indicate contamination following the highest calibration standard, corrective action, such as calibrating with a lower concentration for the highest standard, must be taken and successful analysis of an instrument blank following the highest standard analyzed determines the highest concentration for which carryover does not occur.
Matrix Interferences
Aqueous
Interferences related to the matrix can be caused by the co-extraction of contaminants from the sample. These matrix interferences can have considerable variation from sample to sample. For example, there are problems associated with high total organics content (TOC) in water, which can cause matrix suppression of target analytes and lead to poor recoveries.
Tissue
Interferences related to the matrix can be caused by the co-extraction of bile salts (for example, taurodeoxycholic acid, taurochenodeoxycholic acid, and tauroursodeoxycholic acid) with PFOS from the tissue sample. These bile salts can vary considerably from sample to sample and by species. The use of carbon clean-up steps, such as those required by USEPA Method 1633A, helps eliminate these interferences in most extracts; however, when excessive amounts are present in the extract, these steps may only reduce the amount of these bile salts. To address the potential interference, USEPA Method 1633A requires that the chromatographic conditions are adjusted such that these bile salts elute at a retention time beyond 1 minute from the retention time window of PFOS. Since bile salts can be associated with other matrix types (for example surface waters and wastewaters), the method requires this chromatographic separation to be demonstrated with each analytical sequence, regardless of the matrix type of the sample.
11.2.2 Qualitative Techniques
The Priority Topics for Sampling and Analysis include information about ultrashort-chain PFAS analysis (see Section 1.5.3), consumer product testing (see Section 1.5.4), and field screening tools (see Section 1.5.7).
Several techniques employing indirect measurement have been developed that can more comprehensively assess sites for a wider range of PFAS contaminants than in the typical LC/MS/MS or GC-MS/MS methods. These qualitative techniques are not yet standardized through a published USEPA method and range in commercial availability. To date, these techniques have not undergone multilaboratory validation and tend to be inherently nonquantitative; accordingly, data from these methods may supplement, but not replace, the results of quantitative methods.
11.2.2.1 Overview of Qualitative Techniques
Because of the large number of PFAS and their varied structural characteristics, a single targeted method on either LC/MS/MS or GC-MS/MS may be unable to quantify all PFAS that may be present in a sample. When the release source is well understood and the types of PFAS present are both known and amenable to regular PFAS analysis methods (for example, LC/MS/MS of ionic PFAS or GC-MS/MS analysis of neutral PFAS), a targeted analytical approach may be sufficient to adequately characterize a release. For releases that are not well understood or consist of multiple sources, alternative ways of measuring PFAS in a more comprehensive but less targeted fashion may be desirable. Additionally, PFAS that are in polymeric form, such as those used in coatings for paper and textiles, are not amenable to LC- and GC-based separation techniques; they may also not be effectively extracted, even with rigorous methods.
Five primary techniques have been developed to characterize unknown PFAS in a sample. Only USEPA Method 1621 has been multilaboratory validated but none have been promulgated by USEPA. They are described in more detail in the sections below.
- The total oxidizable precursor (TOP) assay measures PFAA precursors.
- Particle-induced gamma-ray emission (PIGE) spectroscopy measures elemental fluorine isolated on a thin surface.
- USEPA Method 1621, Determination of Adsorbable Organic Fluorine (AOF) in Aqueous Matrices by Combustion Ion Chromatography (CIC) and extractable organic fluorine (EOF), also paired with CIC, measure the organofluorine content of a sample as fluoride on an ion chromatograph.
- High-resolution mass spectrometry techniques, such as quadrupole time-of-flight (qTOF) MS/MS, can tentatively identify PFAS structures through library matching or in-depth data analysis.
- Chemical Ionization Mass Spectrometry (CIMS) technique detects gas-phase PFAS, particularly fluorotelomer alcohols (FTOHs) and other oxygenated PFAS.
11.2.2.2 TOP Assay
Technique Description
The TOP assay converts PFAA precursor compounds to PFAAs through an oxidative digestion. The increase in PFAAs measured after the TOP assay, relative to before, is an estimate of the total concentration of PFAA precursors present in a sample, because not all PFAS present will be subject to quantitation or oxidation, and some will remain as undetected PFAS. The PFAAs generated have perfluoroalkyl chain lengths equal to, or shorter than, the perfluoroalkyl chain lengths present in the precursors (Houtz et al. 2013; Houtz and Sedlak 2012; Weber et al. 2017; Dauchy et al. 2017).
The TOP assay is a technique developed to estimate oxidizable precursors that can transform to PFAA end products that are included in the target analyte list (Houtz et al. 2013; Houtz and Sedlak 2012). A sample is analyzed using conventional LC/MS/MS to determine the baseline levels of PFAAs present in the sample. A separate aliquot of the sample is then exposed to a highly basic persulfate solution and then placed in a sealed container at an elevated temperature (for example, 85°C, in a water bath or other heating device) to thermolyze persulfate into a sulfate radical. At elevated pH, the sulfate radical is scavenged by hydroxide and forms a hydroxyl radical, which then converts the free PFAA precursor compounds to PFAAs. The predominant products (for example, > 95% in control experiments) of the precursors are the perfluoroalkyl carboxylates, whether or not the precursors contain sulfonamido or telomer functionalities. After sufficient time has elapsed to convert all the persulfate, the samples are removed from the heated environment (for example, a water bath), cooled to room temperature, and neutralized prior to LC/MS/MS analysis. The increased concentrations of PFAAs generated after the oxidation step provide an estimate of the concentration of oxidizable PFAA precursors.
The technique can be applied to aqueous (Houtz et al. 2013; Houtz and Sedlak 2012; Houtz et al. 2016; Weber et al. 2017; Dauchy et al. 2017) and solid samples (Houtz et al. 2013). In most cases, samples need to be pretreated prior to oxidation to remove competitive organic compounds. For aqueous samples, dilution may be sufficient, although extraction techniques may be used to further remove matrix effects. Soil samples are extracted prior to persulfate treatment, and the extracts are cleaned with ENVI-Carb prior to treating the evaporated extract. The specific extraction procedure used may affect which PFAA precursors are retained for oxidation. For example, acidic extraction procedures may be required to remove cationic precursor compounds from soils (Barzen-Hanson 2017; Mejia-Avendaño et al. 2017).
Possible Technique Uses
The TOP assay may be used to estimate a total concentration of free PFAA precursors in a sample. In some cases, oxidation can be incomplete (Ventia 2019). The total PFAA precursor or total PFAS concentration is considered conservative for the reasons explained below in Technique Limitations. Because the method depends on a compound containing a perfluoroalkyl group, it is highly specific to PFAS. The chain lengths of the PFAAs generated after oxidation provide an indication of whether the precursors are predominantly short- or long-chained, although the production of a particular Cn (where “n” signifies the number of carbons in the alkyl chain) PFAA is not equivalent to the concentration of PFAA precursors containing the same chain length. However, if significant amounts of PFOA are generated after oxidation, that is an indication that the sample contains a comparable concentration of C8 or longer PFAA precursor compounds.
The TOP assay is the most widely commercially available of the qualitative techniques and is typically accepted as a means of determining PFAS load on remediation media to estimate the replacement cycle, but not for site characterization.
Technique Limitations
As mentioned above, the TOP assay does not differentiate between precursors that contain telomer or sulfonamide functionalities, because all these precursors are chemically oxidized primarily to perfluoroalkyl carboxylates. This is significant because a precursor that would likely form PFOS in the presence of a mixed consortium of aerobic bacteria will convert to PFOA under the conditions of TOP assay. The production of branched perfluoroalkyl carboxylates could be attributed to precursors derived from an ECF-based manufacturing process, but environmental samples may not contain the same distribution of branched and linear isomers as was originally generated from the ECF manufacturing process.
The TOP assay results in a mixture of PFCA products from the conversion of fluorotelomer-based compounds (Houtz and Sedlak 2012). For example, 8:2 FTS was converted to 3% PFNA, 21% PFOA, 27% PFHpA, 19% PFHxA, 12% PFPeA, and 11% PFBA in control experiments. Two limitations of the technique arise because of this effect. First, the production of PFOA, for example, is not equivalent to the C8 precursor concentration, because PFOA can be generated from longer-chain telomer compounds and is only a partial product of C8 telomer products. Second, some shorter-chain PFCA products of telomer compounds are not captured, depending upon the analytical methods used for measurement. For example, only 73% of 6:2 FTS was recovered, from a mass balance standpoint, as PFCA products PFBA and longer in control experiments (Houtz and Sedlak 2012). As a result, the TOP assay may under-quantify short-chain PFAA precursors, including those that are telomer-based. Sulfonamido compounds in control experiments did not exhibit a distribution of products; the Cn precursor forms the Cn PFCA in a 1:1 molar ratio.
Some studies have been published on the effectiveness of the oxidative process of the TOP assay on large molecular weight polymer compounds or newer ether-linked PFAS such as ADONA (Zhang et al. 2019). Because PFAS polymers have shown limited ability to biodegrade (Russell et al. 2008; Russell et al. 2010; Washington et al. 2009) relative to low molecular weight free PFAA precursor compounds (Wang et al. 2005; Lee, D’eon, and Mabury 2010; Wang et al. 2011; Harding-Marjanovic et al. 2015), the TOP assay may be similarly ineffective at converting PFAS polymers to free PFAAs. The TOP assay cannot be used to measure large molecular weight polymeric PFAS unless they are proven to break down in the assay.
For many samples, the TOP assay requires adjustments in dilution, sample preparation, or reagent dosing to achieve complete conversion of PFAA precursors. Standardized quality guidelines are needed to ensure that TOP assay data reflect full conversion of PFAA precursors.
11.2.2.3 PIGE
PIGE is a nondestructive analytical technique that takes advantage of the unique gamma-ray wavelength emission of fluorine when impacted with a proton ion beam. The technique is not compound-specific, but is able to assess total fluorine content of a variety of materials isolated on a thin surface (0.22 mm) (Ritter et al. 2017). The sample is secured in the instrument and bombarded ex vacuo under a 3.4 MeV beam with an intensity of 10 nA for approximately 180 seconds. Two gamma rays characteristic of the decay of the 19F nucleus (110 keV and 197 keV) are measured and the responses integrated. Quantification is achieved with comparison to fluorine-based calibration standards.
In the published literature, PIGE has been used to demonstrate total organofluorine concentrations in papers and textiles (Ritter et al. 2017; Robel et al. 2017) and in food packaging (Schaider et al. 2017). It has also been used on an experimental basis to evaluate organofluorine concentrations in extracted water and soils, but those results are not yet available in the peer-reviewed literature.
Possible Technique Uses
PIGE is a rapid screening technique to measure fluorine on surfaces. If a sample does not contain significant amounts of fluoride or can be prepared to remove inorganic fluoride, PIGE can become a technique specific for organofluorine; however, it is not specific for PFAS. It is a proven way to measure total fluorine in matrices containing high concentration of fluorinated polymeric material, which is a limitation of both the TOP assay and AOF. It also requires relatively minimal sample preparation to analyze fluorine content in commercial products. Sample preparation of environmental samples for PIGE analysis is likely to require a similar level of sample preparation, along with the limitations of extraction techniques, as the TOP assay or AOF.
Technique Limitations
PIGE is not specific to PFAS and, depending on the preparation, it is also not specific to organofluorine. The polymeric compounds that PIGE has been used to detect in consumer products may not contain perfluoroalkyl groups or may not be capable of breaking down to free PFAS. In addition, PIGE cannot distinguish between organic and inorganic fluorine; therefore, the presence of inorganic fluorine can cause results to be biased high.
PIGE also does not provide any differentiation on PFAS perfluoroalkyl chain length present in a sample. Depending on how the sample is prepared prior to the instrumental analysis, samples may be biased toward measurement of long-chain PFAS, as with the TOP assay and AOF.
Extraction methods for PFAS in environmental samples have not yet been demonstrated for this technique. When using SPE to extract environmental aqueous samples prior to PIGE analysis, cartridges that are suitable to hydrophobic and anionic PFAS may not retain positively charged PFAS of interest. For soil samples, the extraction method also determines the PFAS likely retained. However, by using targeted extraction techniques for PFAS in environmental samples, the method becomes much more specific for PFAS.
The range of operating conditions for PIGE has not been standardized and so far, the technique has been demonstrated with only one academic laboratory.
11.2.2.4 Adsorbable Organic Fluorine with Combustion Ion Chromatography
USEPA Method 1621, Determination of Adsorbable Organic Fluorine (AOF) in Aqueous Matrices by Combustion Ion Chromatography (CIC) measures the fluorine content of environmental samples (USEPA 2024). An aqueous sample is passed through a carbon-based sorbent on which the fluorine-containing organics adhere. A nitrate wash of the sorbent is used to eliminate inorganic fluorine. The carbon sorbent is then combusted at high temperatures and the gaseous stream is passed through deionized water, which is then analyzed for fluorine content (as fluoride) by ion chromatography.
The AOF/CIC and EOF/CIC techniques have also been demonstrated on human blood samples (Miyake et al. 2007; Yeung et al. 2008) and various environmental aqueous samples (Miyake et al. 2007; Wagner et al. 2013; Dauchy et al. 2017; Willach, Brauch, and Lange 2016). Presumably, the method could be adapted to other types of matrices to measure organofluorine in soils or biota. The matrices could be extracted for PFAS, the extract resuspended into an aqueous solution that could adhere to the activated carbon, and then analyzed with CIC. As with the TOP assay, the specific extraction procedures would influence whether some or all PFAS are retained and ultimately measured as combusted fluoride product. Alternatively, it is possible that the technique could be used without extraction to directly combust organofluorine-containing products.
Possible Technique Uses
AOF can be used to measure PFAS or other fluorine-containing compounds as an aggregate organofluorine concentration. It can be a screening tool to determine if a significant concentration of fluorine-containing compounds is present in an aqueous sample or other sample from which the organofluorine content can be extracted. A pooled MDL of 1.5 µg f–/L is listed in EPA Method 1621 as achievable by a well-prepared laboratory (USEPA 2024).
Technique Limitations
Like PIGE, AOF is not specific to PFAS. If a sample contains relatively high concentrations of chemical compounds that are not targets of the investigation (for example, fluorine-containing pharmaceuticals), then the organofluorine may be falsely attributed to PFAS content and bias “total PFAS” measurements high.
AOF does not provide any differentiation on PFAS perfluoroalkyl chain length present in a sample. Some short-chain PFAS may be unable to sorb to the activated carbon material that is combusted, but this will depend significantly on laboratory-specific procedures.
Extraction methods for PFAS in commercial products and solid samples, coupled with this technique, have not yet been demonstrated. A high concentration of inorganic fluoride concentrations may be challenging to remove from some matrices and would result in samples biased high for total organofluorine that was actually attributable to fluoride.
Like PIGE, the range of operating conditions for AOF-CIC has not been standardized. In addition to the limitations mentioned above, some matrices may contain sufficient competitive organics or other materials that coat the activated carbon to prevent complete retention of organofluorine compounds.
11.2.2.5 High-Resolution Mass Spectrometry
Technique Description
Quadrupole time-of-flight mass spectrometry (qTOF/MS) can be used to determine both the chemical formula and structure of unknown PFAS in a sample however, analytical standards are required for unequivocal structural identification. More information is available in Section 11.4.3.
High-resolution mass spectrometry has been used to tentatively identify the molecular formulas and structures of unknown PFAS (Newton et al. 2017; Moschet et al. 2017; Barzen-Hanson et al. 2017). Similar to targeted PFAS analysis, techniques such as LC or GC are used to separate compounds in a sample so that individual PFAS can be resolved. The mass is measured using a time-of-flight or other high-resolution detector, and the molecular formula is proposed. If an MS-MS technique is used, the fragments of the precursor compound can be used to piece together the structural arrangement of the compound. To identify compounds that are specifically PFAS, versus other organics present in the sample, compounds with negative mass defects (that is, the accurate mass is slightly less than the nominal mass) can be selected. Fluorine is one of the few elements that has a negative mass defect, and the inclusion of multiple fluorines in a PFAS molecule means that net mass defect of the molecule will likely be negative. Compounds that are either 50 or 100 mass units apart also identify homologous series of PFAS separated by one or two CF2 groups. MS libraries of previously identified PFAS exist for targeted matching, although they will not definitively identify an unknown compound.
Possible Technique Uses
Such high-resolution mass spectrometry analyses of PFAS can tentatively identify the structures of unknown PFAS and can also be used, in comparison with analytical standards of known compounds, to semi-quantitatively estimate their concentrations. Accurate identification of compounds using high-resolution MS is a time-intensive and expensive process. Therefore, a high motivation for knowing the exact PFAS structure, for instance in differentiating forensically between two different sources, may be the biggest driver of its use for PFAS analysis. High-resolution MS is best suited for media in which unknown PFAS are likely to be present in significant concentrations. When many other non-PFAS are present in the sample, the MS signal of competing compounds will likely obscure the signal of PFAS. Sample preparation steps can inadvertently or intentionally select for certain types of PFAS. As user skill and data interpretation time increase, accurate identification of PFAS is likely to improve.
Technique Limitations
High-resolution mass spectrometry cannot definitively identify the exact structure or formulas of PFAS without comparison to reference materials or analytical standards.
Not all PFAS, even if present in a prepared sample, can or will ionize under the conditions to which the instrument is tuned. A skilled instrument operator may be able to adjust the instrument conditions to match the types of compounds expected.
False positives are much more likely to result using high-resolution MS than with the TOP assay, AOF, or PIGE. Compounds may be mistakenly identified as PFAS, and even when correctly identified, their concentrations may be greatly over- or underestimated when other compounds are used for comparative quantitative purposes.
11.2.2.6 Chemical Ionization Mass Spectrometry
Chemical ionization mass spectrometry (CIMS) can be used to detect gas-phase PFAS, particularly fluorotelomer alcohols (FTOHs), and other oxygenated PFAS.
Although GC/MS has only been applied to PFAS analysis in stack emissions with the release of OTM-50, it is also particularly applicable to (ambient) atmospheric samples. Other than OTM-50, there are no published methods for using GC/MS for PFAS analysis, despite the distinct advantages for certain compound classes (for example, fluoro-telomer alcohols) of using GC/MS directly or after derivatization (chemical reaction to convert analyte of interest to a GC/MS-amenable “derivatized compound”) (Langlois et al. 2007), or using large-volume injection (Scott et al. 2006).
11.3 Data Evaluation
Evaluation of data involves looking at all the factors that indicate whether the data are:
- precise (agreement between results that are supposed to be similar)
- accurate (how close they are to the true concentrations)
- representative (results characterize the site properly)
- comparable (data compare well to other data)
- complete (all the samples and compounds requested were reported, especially for critical samples that represent a point of exposure, such as drinking water)
- sensitive (nondetect data reported with concentrations below required regulatory or risk-based level)
These factors are illustrated in Figure 11-1, and guide users through the process of looking at their data (field collection and laboratory information) with a critical eye.
The USEPA has guidance to aid in evaluating PFAS drinking water data generated in accordance with USEPA 537 as well as a technical bulletin to aid in the review of PFAS data generated for all other media (USEPA 2018, 2019). The USDOD EDQW has published PFAS Data Validation Guidelines, for evaluation of PFAS data generated in accordance with the DOD/DOE Quality System Manual (QSM) Table B-24 (USDOD 2022). ASTM has published a Standard Guide for PFAS Data Evaluation (ASTM 2025).

Figure 11-1. Data evaluation factors.
Source: H. Albertus-Benham, Wood Environment & Infrastructure, used with permission.
11.3.1 Pre-sampling Planning
To ensure the usability of the data, communication with the laboratory that is performing the analysis is important. It is incumbent on the data user to collect information about the methodology to be employed by the laboratory. Figure 11-2 contains laboratory considerations related to data usability in order to plan a sampling program.
Figure 11-2. Laboratory planning considerations for data usability.
Source: Modified from figure by H. Albertus-Benham, Wood Environment & Infrastructure. Used with permission.
The most important goal of data usability is to ensure that the PFAS data generated are usable to meet the stated data needs and that the user understands any limitations in the use of the data due to potential uncertainty or bias. Overall usability of data is judged by evaluating the quality of the results compared to the data quality objectives (DQO) of the project. Therefore, establishing these project DQOs and communicating them to the field sampling team and the laboratory prior to sample collection and sample analysis is vital to ensuring that the correct methods, correct compounds, and adequate sensitivity are reported for your samples.
11.3.2 Overall Usability of the Data
Three questions are most important in evaluating data: (1) Have the results exceeded a level of concern?, (2) Do these results make sense?, and (3) Are data of acceptable quality? To judge whether results have exceeded a level of concern, the potential bias or uncertainty in the data should be evaluated along with the sensitivity of the results. At a minimum, it is recommended that a report from the laboratory contains a cover letter (or narrative) explaining sample receipt, analytical methods, and any QC deviations plus data sheets for field samples and QC samples (method blanks, laboratory control samples, sample duplicates, matrix spikes), which should also contain results for sample-specific QC (such as internal standard recoveries). Often the most critical data for a project are the non-detects to prove the absence of compounds of concern at specific concentration levels (quantitation limits). Therefore, before evaluating QC associated with your samples, the data should be evaluated to ensure that all compounds required are reported with quantitation limits at or below the project’s required sensitivity objective. If this sensitivity is not acceptable, then the data may be of very limited use.
If the reported compound list and quantitation limits are acceptable, then the associated QC results (for example, EIS recoveries, results of blanks, laboratory control sample recoveries, etc.) can be compared to project DQOs to evaluate potential uncertainty in the data. The formal systematic process of this QC evaluation is called data review or validation. The approach to data validation is well documented; for example, see the USEPA National Functional Guidelines (USEPA 2020, USEPA 2020), and beyond the scope of this document; however, evaluation of all of the QC associated with the sampling and analysis of a set of samples will lead to an understanding of the uncertainty in the data.
Some critical QC issues might result in unusable data or concern for project actions. For example, if the data are considered biased low based on low QC results and the sample concentrations are at or near the level of concern or an action level, it may be that the true sample concentration exceeded the action level. Conversely, if the sample data are considered biased high based on high QC results and the sample concentrations are near but below the levels of concern or action level, then there is added certainty that the data do not exceed the action levels.
Once the data have been adequately reviewed for accuracy to determine if there are limitations to their use or uncertainties to be considered during use, the results should be evaluated by answering the following questions:
- Do field duplicates, if performed, indicate acceptable precision for the sampling and analysis?
- Do the data from the current sampling event compare well with historical data?
- Do the data make sense from a spatial and temporal point of view?
- Do the data make sense from a spatial point of view from one sampling point to the next across the project area?
This type of review can point out data trends or areas of concern (for example, interferences with project analytes) that could not be elucidated by looking at a single data point and may lead to overall project changes, such as a need to increase sampling density to improve data representativeness, correction of procedures for collecting samples to minimize contamination, changes in methods of analysis to achieve project sensitivity requirements, etc. Following this review, the data user can determine whether the data set is complete and sufficient for project decisions and data uses or whether additional samples need to be collected and analyzed.
11.3.3 Sensitivity
A quantitation limit (QL), or Limit of Quantitation (LOQ) is the limit of accurate quantitation for a specific analyte in a specific sample after any adjustments have been made for sample amount, dilutions, or percent moisture. Typically, the QL or LOQ concentration is selected as the lowest nonzero standard in the calibration curve for each analyte. It considers the sample size, matrix effects, and any dilutions made during the analysis of that particular sample. Because of varying properties between samples, the QL can vary from sample to sample and analyte to analyte. The QL should represent the level at which reliable qualitative and quantitative information is routinely reported (Table 11-3, included in the PFAS Analytical Methods Excel File). When project-specified decision levels or action levels are near the QL, at least one QL check is recommended in all sample batches to demonstrate adequate quantitation at the lowest concentration.
Sensitivity is related to the QL in that sensitivity refers to the capability of a method or instrument to identify a given analyte at a given concentration and reliably quantitate the analyte at that concentration. If a specified analyte is not reported by a laboratory to be in a specified sample, it does not necessarily mean that the chemical is not present; it is an indication that the concentration of the analyte may be below the method sensitivity.
Detected PFAS results between the method detection limit (MDL) and QL (that is, “J” values) can generally be reported if all qualitative identification criteria are achieved. Typical QLs for PFAS are as follows:
- common PFAS analytes in aqueous matrices: 1–16 ng/L (ppt)
- common PFAS analytes in solid matrices: 0.16–1.6 ng/g (ppb)
Sometimes even though lower QLs were planned for, the laboratory may have to perform dilutions, which causes the QLs to be elevated. Ensure that the dilution performed by the laboratory was reasonable. If there are elevated concentrations of specific target analytes or interferences, then the dilution is likely justified and the presence of elevated QLs may not be an issue if these other target analytes are present at very high levels.
If a dilution was performed and it is not obvious why (for example, low concentrations or nondetect results for target analytes), then inquire with the laboratory why the dilution was performed. This could happen due to elevated concentrations of non-target compounds but should be documented.
The QLs can also be affected by the sample preparation parameters, the mass of solid sample or volume of aqueous sample used in the extraction, or the final volume of the extracts. If a complex matrix is encountered, the sample sizes may be reduced and/or the extract volumes may be increased, causing the QL to be elevated accordingly.
11.3.4 Target Analyte Lists
Target analyte lists for PFAS will vary by laboratory and regulatory program. The data user should work with the laboratory to ensure that the correct list is being reported, as dictated by the project objectives. In general, Table 11-4 (included in the PFAS Analytical Methods Excel File) includes the common PFAS reported by existing laboratories. The selected list may be dependent upon project objectives, regulatory requirements, as well as the potential source of PFAS contamination (for example, AFFF, landfill, chromium electroplating). Note that a published method may not include analytes important to characterization of a particular matrix, for example, diPAPs for biosolids characterization (Dickman and Aga 2022).
11.3.5 Linear and Branched Isomers
It is also important to note that PFOS and PFOA (and other PFAS as well) contain a mixture of linear and branched isomers, which can be significant when the laboratory is quantifying these chemicals. Very few standards are available for branched isomers; some standards are for qualitative evaluation and some are quantitative. If branched isomers are not included in the sample quantitation by the lab, the resulting concentrations will be underestimated.
In general, all laboratories should be reporting the sum of the linear and branched isomers for PFHxS, PFOS, PFOA, PFNA, PFOSA, NMeFOSE, NEtFOSE, NMeFOSA, NEtFOSA, NMeFOSAA, and NEtFOSAA because these are the PFAS for which both linear and branched analytical standards are available as of this publication. In the absence of a standard that includes branched isomers, only the peak associated with the linear isomer is integrated. As more analytical standards become available, more PFAS should be reported as linear and branched in the future.
Figure 11-3 shows an example of the integration performed correctly and incorrectly. It is the responsibility of the data user to ensure that the laboratory is performing the integration of the target PFAS to include both linear and branched isomers. This requires upfront communication with the laboratory and a possible independent review of the laboratory raw data by a qualified chemist/data user to verify the integrations were properly performed.
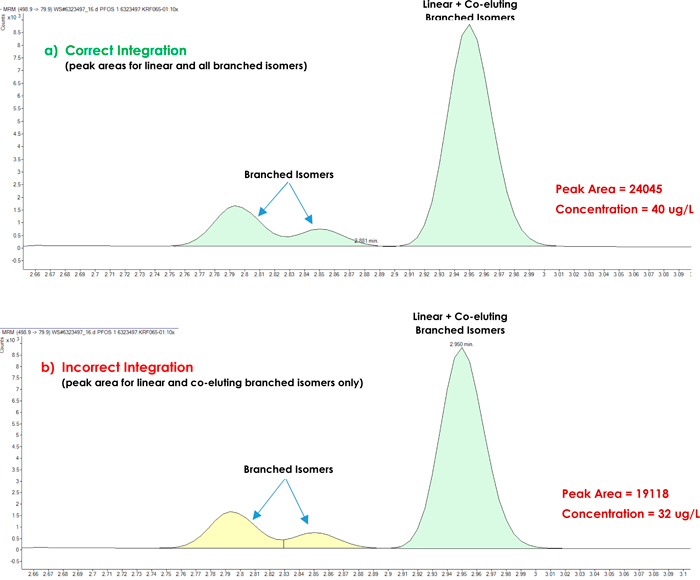
Figure 11-3. LC/MS/MS data illustrating a) complete integration of linear and branched PFOS, and b) partial integration of PFOS. Discrepancies in concentration will depend on the fraction of branched versus linear PFAS present, but in the current example PFOS concentrations in b) were 20% lower than in a).
Source: Bureau Veritas Laboratories, Mississauga, Ontario, Canada. Used with permission.
11.3.6 Isotope Dilution Standard Results and Surrogates
Isotope Dilution Standards
The isotope dilution technique involves quantitation of a compound of interest using a labeled isotope of that very compound. A variety of isotopically labeled analogs (for example, carbon-13 analogs of the compounds of interest) are added to a sample prior to extraction or prior to analysis when extraction is not required. These isotopically labeled analogs are sometimes referred to as extracted internal standards, as defined in USEPA Method 1633A, or isotope dilution analogues, as defined in USEPA Method 533, and function from a data usability standpoint as both an internal standard (used in the calculation of the target compounds) and as a surrogate standard (calculation of the recovery of the standard). Ideally, the number of isotopically labeled analogs used in the isotope dilution technique matches the number of target compounds. Non-extracted internal standards, as defined by USEPA Method 1633A, or isotope performance standards, as defined by USEPA Method 533, are also added to the sample or extract immediately preceding analysis and are used to calculate recovery of the extracted internal standards.
For the isotope dilution methods, quantitation of the target compounds is performed relative to the response of the isotopically labeled analog, which should recover in a manner similar to how the nonlabeled compounds recover. Effectively, the sample data are recovery-corrected for losses that might have occurred during sample processing. The isotope dilution recovery correction procedure greatly improves the accuracy of the analysis and is an improvement over other techniques for the analysis of complex samples for analytes requiring high sensitivity. Chemical standards manufacturers are working to make a wider variety of labeled isotope compounds available to further improve accuracy of the methods for all compounds under investigation (for example, fluorotelomers, precursors, various isomers of carboxylates and sulfonates).
Acceptance criteria or control limits for isotopically labeled analog recoveries are either developed by the laboratory or dictated by the requirements of the project (for example, work plan or QAPP-specified criteria, regulatory criteria, or method criteria). Poor recovery of EIS in complex matrices is common; however, if a project requires ongoing analysis of a problematic matrix, the laboratory should perform method development to improve recovery, if possible (for example, change in cleanup procedures, change in the transition ions monitored, etc.). If EIS recovery is very low (for example, < 10% recovery), nondetections associated with the EIS may be false negatives and may not be useable for project objectives. If EIS recovery is low but ≥10%, there may be an indeterminate bias for the affected PFAS. If EIS recovery is high, there will be no effect on non-detects, but positive results for PFAS may have an indeterminate bias.
In the case where an isotope dilution extract is analyzed and requires re-analysis at a high dilution, the sample extract may need to be refortified with labeled isotope compounds or, if possible, a smaller aliquot of sample may need to be extracted to obtain adequate responses of EISs. In reporting the final data, the isotope recovery results from the initial analysis should not be used to adjust the data from the secondary dilution analysis because these recoveries may be affected by ion suppression or ion enhancement due to the elevated concentrations of target PFAS and therefore may not be reflective of the extraction efficiency or other matrix interferences. The result from this scenario is no longer quantitated from an isotope dilution but is calculated from an internal standard calculation and should be noted as such in the case narrative.
Surrogates in Non-Isotope Dilution Procedures
Method 537.1 uses four surrogates for 18 target compounds, while EPA Method 8327 uses 19 surrogates for 24 target compounds. Injection internal standards are also added to the sample extract immediately preceding analysis. Quantitation of the target compounds and surrogates is performed relative to these injection internal standards. The results from the non-isotope dilution technique report concentrations of the target compounds and recovery results for surrogates, and it is up to the data user to determine the impact (that is, bias) of the extraction and analysis on the sample results because results are not recovery-corrected.
Acceptance criteria or control limits for surrogate recoveries are either developed by the laboratory or dictated by the requirements of the project (for example, work plan or QAPP-specified criteria, regulatory criteria, or method criteria). Poor recovery of surrogates in complex matrices is common; however, if a project requires ongoing analysis of a problematic matrix, the laboratory should perform method development to improve recovery, if possible (for example, change in cleanup procedures, change in the transition ions monitored, etc.). If the recovery for a surrogate is below criteria, compounds associated with this surrogate may be biased low. If surrogate recovery is very low (for example, < 10% recovery), nondetections associated with the surrogate may be false negatives and may not be useable for project objectives. If surrogate recovery is high, there will be no effect on non-detects but positive results for PFAS may be biased high.
11.3.7 Blank Contamination
As a consequence of the extensive use of PFAS, samples that may not contain PFAS can become contaminated if they come into contact with samples or materials containing PFAS. The types of blanks commonly used to evaluate contamination are field-based blanks and lab-based blanks. Field-based blanks include field reagent blank (field blank), source water blank, and equipment rinse blank. Laboratory-based blanks include method blank, lab reagent blank, and instrument blank. Reagent, source, field, and method blanks are prepared and analyzed using the same procedures as for the field samples. Instrument blanks are analyzed periodically to verify the instrument is clean for analysis of subsequent samples.
The lab reagent blank is used to evaluate the potential PFAS contamination from the reagent water source used to generate the field-based and laboratory-based blanks. A systematic review of all of the blank results compared to the associated field sample results (the group of samples associated with the field-based and lab-based blanks, or the analytical batch of samples associated with a specific method blank) must be made to determine whether the field sample results are accurate. For example, if the reagent water source used in the field is nondetect for PFAS, but contamination is found in any of the other field-based blanks, this indicates potential contamination of the associated field samples from the sample bottle itself and/or during collection, handling, or transport to the laboratory. However, if a laboratory-based blank is also contaminated, the contamination observed in the field-based blanks may have been due to sample handling at the laboratory.
If the conclusion of this systematic blank data review is that an associated sample result may have been contaminated, then the sample result is considered to be biased high or may be a false positive, depending on the magnitude of the blank contamination compared to the field sample result. A general rule of thumb is that if a sample contains a contaminant within 5x–10x the concentration in the associated blank, the results may be biased high or result in a false positive in the sample (USDOD 2019).
11.3.8 Duplicate Results
Laboratory replicates are two separate aliquots of the same sample prepared at the laboratory and put through the entire sample preparation and analytical procedures. Field duplicates are two separate samples collected at the same location at the same date and time that are prepared and analyzed in the same manner. Laboratory replicates may be performed in lieu of an LCS duplicate or MSD. It should be noted that sometimes laboratories report the results of laboratory replicates performed on samples that are from a different project (that is, batch QC); if the laboratory replicates reported are not from a sample at the site of interest, then these results should not be used in the evaluation of sample data.
During data evaluation, the relative percent difference of each detected analyte versus the acceptance limits should be reviewed. The acceptance limits should be provided within the laboratory report and are either regulatory- or method-specific. When both results are < 2x the QL, the potential uncertainty increases, and therefore, the acceptance criteria may need to be adjusted.
- If both results are < 2x the QL, relative percent difference criteria can be doubled.
- If one result is detected and one result is not detected, then the evaluation will depend on whether the detected result is > 2x the QL or not. If one result is > 2x the QL and the other result is nondetect, then the variability is considered unacceptable and there may be potential uncertainty in the results for this sample.
Variability in laboratory replicate and field duplicate analyses could be from the sampling process, such as inefficient homogenization procedure in the field. It could also be from the laboratory aliquoting process, or it could be due to heterogeneity in the sample matrix. The effect on project objectives will depend on the screening criteria and how far above or below these criteria the results are. If the results are close to the criteria with significant variability, this may require collection of more samples to better represent the location. If results are significantly above or below the screening criteria with high variability, it may not adversely affect the ultimate decision-making process.
11.3.9 Acid Versus Anion Form of PFAAs
The data user should be aware of the form of PFAS the laboratory is reporting when comparing to project screening criteria. PFAS are typically formulated as acids, but they are present in the environment and in humans in the anionic form. The differences in names used are the result of the different names for the acid form and the anion form of the chemical (see also Section 2.2.3.1). For example, when perfluorobutanoic acid (PFBA) disassociates and loses its hydrogen in water, it becomes the anionic form (perfluorobutanoate). This becomes more important when looking at physical and chemical properties of these chemicals, because whether they exist as an acid, an anion, or a salt will affect how they behave in the environment. Typically, laboratories are reporting the acid form of the perfluorocarboxylic acids and perfluorosulfonic acids.
Some target PFAS, such as PFHxS and PFOS, are not available as acids, but rather as their corresponding salts (for example, K+, Na+, and Li+). These salts are acceptable starting materials for the stock standards, provided the measured mass is corrected for the salt content according to the equation below. Note that this correction will result in a minimal change to the mass of the acid but still should be performed for consistency and comparability with other results to ensure the data user that the correct form of PFAS is represented in the final concentration.
massacid = measured masssalt * (MWacid/MWsalt)
MWacid = molecular weight of PFAA
MWsalt = molecular weight of purchased salt
CAS numbers will change depending on if the acid, anion, or salt form of the PFAS is reported (Table 11-7).
Table 11-7. Example of CAS number differences between acid and anion
| Chemical | CAS number |
|---|---|
| PFOA: Perfluorooctanoate (anion) | 45285-51-6 |
| PFOA: Perfluorooctanoic acid (acid) | 335-67-1 |
11.4 Source Identification
As discussed in Section 10.5 and Section 1.3.7, one area of focus for PFAS investigations has been environmental forensics, specifically source identification. With so many industrial processes and transport pathways from which PFAS contamination can originate, attributing the occurrence of one or more PFAS to a particular source is of growing interest.
Source identification relies on the compilation of multiple lines of evidence from analytical data and site information, and the use of uni- and multivariate statistical analyses. Analytical lines of evidence include:
- Extended Target Lists
- Linear/Branched Speciation
- Non-Targeted Analysis (NTA)
These methods are briefly described in this section.
11.4.1 Extended Target Lists
Many commercial laboratories now analyze for more than 40 individual PFAS and some of these compounds may be indicative of a particular product or industrial process. Analyzing samples taken from or near these possible sources and comparing the profiles of those results to the area of concern, also known as chemical fingerprinting, may provide a demonstration of comparability.
11.4.2 Linear/Branched Speciation
The separate reporting of branched and linear PFAS can provide some indication of the process used to synthesize the PFAS detected. Electrochemical fluorination (ECF) produces a larger number of branched isomers than fluorotelomerization, which may be useful in differentiating sources (Benskin, DeSilva, and Martin 2010). Separate reporting of branched and linear PFAS is not typically the default laboratory reporting and should be requested in advance. However, due to isomer-specific differences in instrument response, and fate, transport and organism uptake/depuration, the use of speciated data for any inference more than presence/absence of a manufacturing source type is challenging.
11.4.3 Non Targeted Analysis (NTA)
The ability to identify compounds not targeted for measurement by existing methods is an important need. NTAs are critical to being able to identify these compounds. With NTAs, chromatography (liquid and gas) is combined with high resolution mass spectrometry and multiple ionization techniques to determine atomic molecular weight and associated fragments. These results can be compared to databases for tentative identification (Liu et al. 2019). Further spectral interpretation may result in structural identification. This is particularly useful where no chemical standards exist.
NTA includes high-resolution mass spectrometry and suspect screening and is discussed in more detail below.
11.4.3.1 High-Resolution Mass Spectrometry for Nontargeted Analysis
Although not widely commercially available, recent source research has focused on the use of high-resolution mass spectrometry (HRMS) for more comprehensive qualitative determination and fingerprinting for source attribution. See additional information in Section 11.2.2.5.
Many PFAS exist with no certified reference standard to make an initial calibration plot. As such, non-targeted PFAS may be present in samples that will not be identified or reported on a standard target analyte list, unless requested. HRMS is emerging as a means for discovery and reporting of non-targeted PFAS where each unknown peak may be tentatively identified and quantified at estimated concentrations. Non-targeted analytes can be reported by the identification of fragmented organic molecular ions (m/z) that are matched to an analytical reference library. The reporting includes detection of homologous series with or without the presence of nonfluorinated functional groups that create mass defects. Mass defect is the difference between the nominal and exact mass of an atom, which allows prediction of molecular formulas and is the key advantage to using HRMS for NTA. Liu et al. (2019) categorized PFAS into the main classes of:
-
- Per/polyfluoroalkyl carboxylic acids
- Per/polyfluoroalkyl sulfonic acids
- Fluorotelomers and per/polyfluoroalkyl sulfonamides
- Per/polyfluoroalkyl phosphates
- Per/polyfluoroalkyl alcohols
- Per/poly-fluoroalkylamides
- Per/polyfluoroalkyl sulfates
- N-per/polyfluoroalkyls
Tables are available on the NORMAN Suspect List Exchange (https://www.norman-network.com/?q=node/236). Charbonnet et al. (2022) defined the basic homologue series by CnF2n+1, where n > 2 with isomeric PFAS of the same chemical formula differentiated as structural isomers that may be linear, branched in the perfluoroalkyl tail or the head group, or have varying functional groups that could create mass defect.
As evidenced by SERDP-ESTCP funded projects listed below, the development of mass spectral libraries to match non-targeted analytes to source profiles is part of ongoing research. NTA uses HRMS such as time-of-flight (TOF), ion-trap, or Fourier transform ion cyclotron resonance (FT-ICR) to generate high resolution accurate mass data. As extensive data sets are generated using HRMS, informed data filtering approaches are used to filter the data specific to PFAS related analytes. The data are first screened against previously generated suspect screening libraries that contain chromatographic/spectrometric information for PFAS characteristic to sources such as AFFF formulation, industrial process, and/or manufactured products. Then, mass spectrometry-specific data analyses, such as Kendrick mass defect plots, and general uni- and multivariate statistical analyses, are used to attempt source identification based on the presence/relative abundance of PFAS identified against the suspect screening libraries and other information (Benotti et al. 2020; Charbonnet et al. 2021).
The Interstate Technology and Regulatory Council (ITRC) Contaminants of Emerging Concern (CEC) Team fact sheet on Adoption of Analytical Methods for Identifying CEC provides additional details on non-targeted analyses, including mass spectral libraries used and determining the confidence in compound identifications (ITRC 2023).
Secondary Sources
SERDP PFAS Novel Methods for PFAS Source Tracking and Allocations
ER20–1375 Comprehensive Forensic Approach for Source Allocation of Poly- and Perfluoroalkyl Substances – Chris Higgins, Colorado School of Mines
ER20–1121 Establishing an Approach to PFAS Forensics and a PFAS Source Materials Forensic Library – Mark Benotti, NewFields Government Services
ER20–1205 Machine Learning Pattern Recognition for Forensic Analysis of Detected Per- and Polyfluoroalkyl Substances in Environmental Samples – Tohren Kibbey, University of Oklahoma
ER20–1265 Ultrahigh-Resolution Fourier-Transform Ion Cyclotron Resonance Mass Spectrometry for Fingerprinting, Source Tracking, and Allocation of Per- and Polyfluoroalkyl Substances (PFAS) – Jens Blotevogel, Colorado State University
ER20–1056 Improving Access and Utility of Analytical Data for the Confident Discovery, Identification, and Source-Attribution of PFAS in Environmental Matrices – Benjamin Place, NIST, Department of Commerce
–
Updated January 2026.